Kaposi-Sarkom
Das Kaposi-Sarkom (KS) ist ein angioproliferativer Tumor, der mit dem humanen Herpesvirus Typ 8 (HHV-8) assoziiert ist.1,2 Es wurden vier Varianten beschrieben (Tabelle 1).
Epidemiologie und klinische Merkmale der verschiedenen Typen des Kaposi-Sarkoms.
| Klassisches KS | Endemisches KS | HIV-assoziiertes Kaposi-Sarkom | Immunosuppressions-Assoziiertes Kaposi-Sarkom | |
|---|---|---|---|---|
| Alter | >60 Jahre | 30-45 Jahre | 20-50 Jahre | 60 Jahre |
| Risikogruppen | Mediterrane und jüdische Abstammung | Afrikanische Bevölkerung | HIV-Infektion und MSM | Transplantationsempfänger, Autoimmunerkrankungen |
| Ort | Beine | Beine | Kephalisch, oral, viszeral | Gliedmaßen |
| Extrakutane Manifestationen | Häufig | Oft lymphoadenopathisch | Häufig | Möglich |
| Ausgang | Indolenter Verlauf | Progressiv | Aggressiv, kann mit antiretroviraler Therapie verzögert werden | Variabel, kann sich mit verminderter Immunsuppression zurückbilden |
Abkürzungen: MSM, Männer, die Sex mit Männern haben; KS, Kaposi-Sarkom.
Klassisches Kaposi-Sarkom. Es handelt sich um einen seltenen Tumor, der tendenziell Männer1,3 im mediterranen oder mitteleuropäischen Raum betrifft, mit einer Inzidenz zwischen 0,18 und 13,2 Fällen/Million.4 Es tritt häufiger bei Männern mit chronischen Beinödemen, Diabetes mellitus und Kortikosteroid-Anwendern auf. Die Läsionen präsentieren sich als einzelne oder multiple, langsam wachsende erythematös-violette Plaques oder Knötchen, in seltenen Fällen in Verbindung mit Lymphödemen in den Extremitäten und gastrointestinaler und Lymphknotenbeteiligung. Es hat einen indolenten klinischen Verlauf, und 2 % der Patienten sterben an einer disseminierten Erkrankung.
Endemisches Kaposi-Sarkom. Diese Variante wurde aus dem äquatorialen Afrika berichtet und betrifft junge Erwachsene und vorpubertäre Personen. Bei Erwachsenen nimmt die Variante einen indolenten oder aggressiven Verlauf mit Beteiligung von subkutanem und Knochengewebe, während sie sich bei Kindern in einer aggressiven Form mit generalisiertem Lymphknotenbefall, Beteiligung innerer Organe und fehlenden (oder begrenzten) Hautläsionen manifestiert.5
Immunsuppressions-assoziiertes Kaposi-Sarkom (iatrogene Variante). Diese Variante wurde bei Patienten in Behandlung mit Immunsuppressiva und insbesondere bei Transplantatempfängern berichtet. Das Risiko, ein KS zu entwickeln, ist schätzungsweise 150 bis 200 Mal höher als in der Allgemeinbevölkerung, mit einer mittleren Zeit bis zum Auftreten von 18 Monaten.6
HIV-assoziiertes Kaposi-Sarkom (epidemische Variante). Diese Variante wurde bei Männern mit HIV-Infektion, die Sex mit Männern haben (MSM-HIV+), berichtet. Vor der Ära der hochaktiven antiretroviralen Therapie (HAART) wurde berechnet, dass 25 % der MSM-HIV+ ein KS entwickeln würden, obwohl dieser Prozentsatz progressiv abgenommen hat2,7 (Abb. 1 und 2). Bei diesen Patienten kann es zu einer Beteiligung von Haut und Schleimhaut, Lymphknoten, Magen-Darm-Trakt, Lunge, Milz und Leber kommen.7 KS bei MSM-HIV- wurde ebenfalls berichtet, in diesem Fall mit einem indolenten Verlauf.8
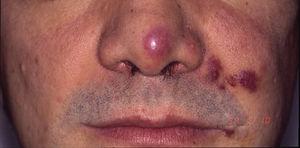
Kaposi-Sarkom bei einem Patienten mit erworbenem Immundefektsyndrom. Erythematös-violette Plaques an der Nasenspitze, dem Mundwinkel und der linken Wange.
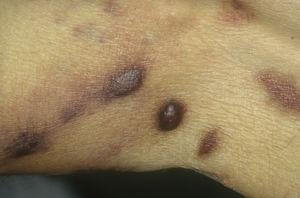
Klassisches Kaposi-Sarkom an den Beinen. Erythematöse bräunliche Plaques am Bein.
Diagnose
Die Diagnose wird klinisch gestellt, aber es wird empfohlen, eine Bestätigung durch eine Biopsie zu erhalten. Die histologische Untersuchung zeigt Spindelzellen, Proliferation von unregelmäßigen Gefäßen mit schlitzartigen Formen, Extravasation von roten Blutkörperchen und leukozytäres Infiltrat mit Plasmazellen und intra- und extrazellulären Hyalinkügelchen in der gesamten Dicke der Dermis sowie das sogenannte Promontoriumszeichen (Abb. 3). Polymerase-Kettenreaktion und immunhistochemische Färbung für das Latenz-assoziierte nukleäre Antigen (LANA-1) des HHV-8-Virus sind positiv.1
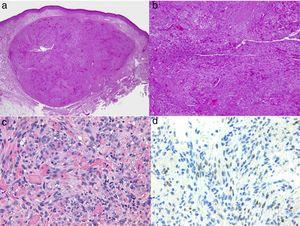
Histologie des Kaposi-Sarkoms.
A, Niedrigvergrößerungsansicht des Kaposi-Sarkoms in knotiger Phase. Gut definierter dermaler Knoten.
B, Hochzellige Läsion mit einigen Lücken in Form von Rissen.
C, Detail von Spindelzellen und roten Blutkörperchen innerhalb der kleinen Gefäße.
D, Immunhistochemische nukleäre Positivität für HHV-8, spezifisch für das Kaposi-Sarkom.
Staging und ergänzende Tests
Klassisches Kaposi-Sarkom. Angesichts der klinischen Präsentation (Alter, lokale Beteiligung, seltener Befall innerer Organe und indolenter Verlauf) ist eine Untersuchung der Haut und der Lymphknoten ausreichend. Ergänzende Untersuchungen werden durchgeführt, wenn der Patient Symptome einer viszeralen Beeinträchtigung aufweist.5
Immunosuppressions-assoziiertes Kaposi-Sarkom. Wie für das HIV-assoziierte KS, das weiter unten besprochen wird, gibt es auch für das Immunsuppressions-assoziierte KS kein Konsens-Staging. Angesichts des fehlenden Konsenses sind die Tests und empfohlenen anwendbaren Kriterien in der Regel die gleichen wie für das HIV-assoziierte KS.
HIV-assoziiertes Kaposi-Sarkom Für diese Variante gibt es kein anerkanntes Staging-System. Eine Röntgenaufnahme des Brustkorbs wird empfohlen, und bei Auffälligkeiten, die auf eine Beeinträchtigung der Atemwege hindeuten, sollte eine Bronchoskopie oder eine Computertomographie des Brustkorbs durchgeführt werden. Es wird auch empfohlen, fäkales okkultes Blut auszuschließen; wenn dieser Test positiv ist, sollte eine Endoskopie des Verdauungstrakts durchgeführt werden.
Im Jahr 1989 schlug das AIDS Clinical Trials Group Oncology Committee eine Stadieneinteilung vor (Tabelle 2), die auf der Ausdehnung der Krankheit, der CD4T-Zellzahl und der systemischen Beeinträchtigung (opportunistische Infektionen, B-Symptome wie Fieber, Gewichtsverlust oder anhaltende Diarrhöe und Karnofsky-Leistungsstatus unter 70 Punkten) basiert. Eine prospektive Analyse zeigte, dass diese Variablen mit dem Überleben der Patienten zusammenhingen, wobei Faktoren, die mit einer guten Prognose assoziiert waren, eine begrenzte Erkrankung, eine CD4-Lymphozytenzahl von mehr als 150 Zellen/mm3 und das Fehlen einer systemischen Beeinträchtigung waren.9
Vom AIDS Clinical Trials Groups Oncology Committee vorgeschlagenes Staging.
| Gute Prognose (0) | Schlechte Prognose (1) | |
|---|---|---|
| Tumor | Tumor nur in der Haut oder in Lymphknoten gefunden, oder minimale orale Beteiligung | Tumor, der eine Ulzeration verursacht Schwere orale Beteiligung KS-Läsionen im Gastrointestinaltrakt KS-Läsionen an inneren Organen außer Lymphknoten |
| Immunität | CD4 ≥ 200/μL | CD4 200/μL |
| Systemische Erkrankungen | Keine opportunistischen Infektionen oder Soor Keine B-Symptome Karnofsky-Performance-Status ≥ 70 |
Opportunistische Infektionen oder Soor B-Symptome Karnofsky-Performance-Status 70 Weitere HIV-assoziierte Erkrankungen (z.B., neurologische Erkrankung oder Lymphom) |
Abkürzung; KS, Kaposi-Sarkom.
Längeres Fieber, Nachtschweiß, mehr als 10 % Gewichtsverlust oder Durchfall, der länger als 15 Tage anhält.
Diese Vorschläge für die Stadieneinteilung wurden gemacht, bevor HAART verfügbar war, und beziehen die Viruslast nicht mit ein, so dass die Anwendung dieses Stadieneinteilungssystems auf klinische Studien beschränkt ist.
BehandlungKlassisches Kaposi-Sarkom
Es gibt nur wenige vergleichende klinische Studien zu den verschiedenen Behandlungen des klassischen KS. In der Regel werden die gleichen Medikamente und Schemata wie beim epidemischen KS verwendet (Tabelle 3).
-
Solitäre Läsionen.
- –
Klinische Beobachtung. Angesichts des Alters der Patienten und der geringen Mortalität kann eine Nachbeobachtung ohne Behandlung eine Option sein. Im Falle eines Lymphödems wird eine elastische Kompression empfohlen.10
- –
Lokale Strahlentherapie. Niederenergetische Strahlentherapie (100kV: 8Gy in einer einzigen Anwendung oder 20-30Gy als fraktionierte Dosis) ist bei isolierten Läsionen wirksam.10 In einer retrospektiven Analyse von 68 Patienten, die mit Strahlentherapie behandelt wurden, wurde bei 85% der Patienten ein gutes Ansprechen beobachtet, mit vollständigem Ansprechen in 58% und Verbesserung der Symptome in 95%.3
- –
Chirurgische Exzision. Bei Läsionen, die Beschwerden verursachen, wie z.B. im Bereich der Akren, wird eine Operation empfohlen. In einer Studie mit chirurgisch exzidierten Läsionen waren die Patienten im Mittel 15 Monate nach der Behandlung rezidivfrei.3
- –
Kryotherapie. Die Kryotherapie kann bei solitären Läsionen mit einer Größe von ≤1 cm, insbesondere im Bereich der Akren, mit einer Anwendungsdauer von 30 bis 60 Sekunden eingesetzt werden. Sowohl die Operation als auch die Kryotherapie haben den Vorteil, dass sie mit guten Ergebnissen wiederholt werden können.
- –
Intralesionaltherapie. Intralesionale Behandlung mit Vinblastin (0,2mg/mL alle 2 Wochen), Vincristin (0,03-0,08mg) oder Interferon alfa (3-5MIU/3 mal wöchentlich für 4-5 Wochen),10 topische Anwendung von Alitretinoin 0,1% Gel (makuläre Läsionen) oder Imiquimod 5% Creme (3 mal wöchentlich für 24 Wochen)10 sind Behandlungen, die in der Literatur empfohlen werden, obwohl es nur begrenzte Erfahrungen mit diesen Therapien gibt.5
-
Disseminierte Erkrankung
- –
Pegyliertes liposomales Doxorubicin (PLD) (20mg/m2 alle 3Wochen). PLD ist die Behandlung der Wahl, außer bei Patienten mit Herzerkrankungen. Das Ansprechen ist gut oder ausgezeichnet, mit partiellem Ansprechen (50 % Rückgang der Anzahl der Tumorläsionen) oder komplettem Ansprechen über 25 Monate bei mehr als 70 % der Patienten.11 Die Dauer der Chemotherapie ist nicht gut etabliert, obwohl empfohlen wird, die Behandlung für 1-2 Zyklen nach dem klinischen Ansprechen fortzusetzen. Die Behandlung mit PLD ist im Allgemeinen gut verträglich, mit begrenzten Nebenwirkungen, und sie ist weniger kardiotoxisch als die Behandlung mit herkömmlichen Substanzen. Daher können höhere kumulative Dosen und längere Behandlungen verabreicht werden. Die schwerwiegendsten Toxizitäten (Grad 3 und 4) sind selten und umfassen Neutropenie und Anämie.12
- –
Vinblastin (3mg/m2/Woche/intravenös oder 6mg/m2/2wöchentlich/iv). Vinblastin bietet gute Ergebnisse, mit Ansprechraten zwischen 50 % und 90 %, obwohl Leukopenie auftreten kann.3,13
- –
Andere Chemotherapien, die mit hohen Ansprechraten assoziiert sind, aber auch unerwünschte Wirkungen haben, sind Paclitaxel iv (100mg/Woche), Bleomycin (15U/Woche für3Wochen und dann alle 3Wochen/intramuskulär) und orales Etoposid (100mg/Tag, 3-5 Tage pro Woche).10 Es gibt nur 1 randomisierte klinische Studie, die Vinblastin iv mit oralem Etoposid verglichen hat; es wurden keine signifikanten Unterschiede im Ansprechen oder Überleben gefunden, aber ein höherer Anteil an unerwünschten Wirkungen wurde bei der Etoposidbehandlung berichtet.13
Algorithmus zur Behandlung des Kaposi-Sarkoms.
| Variante | Behandlung | ||
|---|---|---|---|
| Klassisches KS | Behandlung der Wahl | Andere Alternativen | |
| I. Solitäre oder isolierte Läsionen | a. Nur klinische Beobachtung in Betracht ziehen. b. Lokale Strahlentherapie oder chirurgische Exzision |
Intralesionale Therapie | |
| II. Disseminierte Erkrankung | Liposomales Doxorubicin | Vinblastin, Paclitaxel, Etoposid | |
| HIV-assoziiertes Kaposi-Sarkom | I. Solitäre oder isolierte Läsionen | Initiieren Sie HAART mit oder ohne lokale Therapie (Radiotherapie, chirurgische Exzision oder Kryotherapie) | HAART und intraläsionale Therapie |
| II. Disseminierte Erkrankung | HAART und liposomales Doxorubicin | HAART mit Paclitaxel | |
| Iatrogenes KS (assoziiert mit Immunsuppression) | Aussetzen oder Reduzieren der Dosis des Immunsuppressivums | Befolgen Sie das Schema zur Behandlung von HIV-assoziierten KS | |
Abkürzungen: HAART, hochaktive antiretrovirale Therapie; KS, Kaposi-Sarkom
HIV-assoziiertes Kaposi-Sarkom
-
Solitäre oder begrenzte Läsionen
- –
HAART in Monotherapie oder in Verbindung mit lokaler Therapie (Abb. 4). HAART wird als Erstbehandlung verabreicht, da solche Mittel nachweislich die Immunfunktion wiederherstellen und die Inzidenz und den Schweregrad des Sarkoms senken,2,7 mit Reduktion oder Verschwinden der Läsionen. Bei symptomatischen und unästhetischen Läsionen sind eine Operation,14 eine Elektrokoagulation oder eine Kryotherapie Optionen. Intralesionales Vinblastin15 (0,2 bis 0,3mg/mL, Verabreichung von 0,1mL pro 0,5cm2 Läsion) oder niederenergetische Strahlentherapie (100kV, Dosen zwischen 8Gy/1Fraktion oder 30Gy/10Fraktionen, mehr als 95% komplettes klinisches Ansprechen) können in Betracht gezogen werden.16 Das läsionsfreie Überleben nach 5 Jahren nach HAART-Behandlung, mit oder ohne lokale Therapie, betrug 92% in einer Serie von mehr als 400 Fällen.14
 Abbildung 4.
Abbildung 4.Behandlungsalgorithmus für das HIV-assoziierte Kaposi-Sarkom.
Abkürzungen: HAART, hochaktive antiretrovirale Therapie; IRS, Immunrekonstitutionssyndrom.
Abgeleitet von der Consensus Group for treatment of HIV-associated Kaposi sarcoma. Consensus Meeting. Barcelona: Saned; 1998.
(0.12MB). -
Disseminierte Erkrankung
Eine systemische Behandlung wird bei Patienten empfohlen, die mit HAART behandelt werden und eine ausgedehnte Hautbeteiligung aufweisen (mehr als 15 bis 25 Läsionen), starken Schwellungen, kutanem KS, das auf eine lokale Therapie nicht angesprochen hat oder schnell fortschreitet, KS in Verbindung mit einem Immunrekonstitutionssyndrom oder symptomatischer Beteiligung innerer Organe.
- –
PLD (20mg/m2 alle 3Wochen). HAART und PLD sollten gleichzeitig begonnen werden,17 da die Kombination wirksamer ist als HAART allein.18 In Abhängigkeit vom klinischen Ansprechen werden üblicherweise mehrere Behandlungskurse verabreicht. Ein vollständiges/teilweises Ansprechen wird mit der Kombinationstherapie in 80 % erreicht,19 mit einer 5-Jahres-Überlebensrate von mehr als 85 %. Rückfälle sind begrenzt (13%) und treten im ersten Jahr nach Beendigung der Behandlung auf.20 Das Ansprechen auf PLD ist höher als bei der Kombination aus Bleomycin, Vincristin oder Vinblastin und nicht liposomalem Doxorubicin,21 und liposomalem Daunorubicin.22
- –
Paclitaxel (100mg/m2 alle 2 Wochen). Paclitaxel ist eine Zweitlinientherapie mit günstigem Ansprechen bei 71% der Patienten,23 aber mit niedrigeren Überlebensraten als PLD und höheren Raten von Grad 3-5 Toxizitäten.14,24 Eine Prämedikation mit Dexamethason ist erforderlich und es können schwere Wechselwirkungen mit antiretroviralen Wirkstoffen auftreten.
- –
Andere Therapien. Andere Medikamente wie Etoposid, Vinorelbin, Interleukin12, Bevacizumab und Imatinib wurden ebenfalls eingesetzt, die Erfahrungen sind jedoch begrenzt.5
Immunsuppressions-assoziiertes Kaposi-Sarkom
- –
Setzen Sie das Immunsuppressivum ab oder reduzieren Sie die Dosis. Das Aussetzen oder die Reduzierung der Immunsuppressiva-Dosis kann Spontanremissionen auslösen. Wenn dies unwirksam oder nicht praktikabel ist, werden in der Regel die bei HIV-assoziiertem KS verwendeten Ansätze angewandt.5
Schlussfolgerungen
KS ist ein angioproliferativer Tumor mit verschiedenen Subtypen, die mit fortgeschrittenem Alter, bestimmten afrikanischen Bevölkerungsgruppen, iatrogener Immunsuppression oder HIV-Infektion assoziiert sind. Obwohl HAART zu einem dramatischen Rückgang der Inzidenz und des Schweregrads von KS bei HIV-Infizierten geführt hat, ist es wichtig, sich der unterschiedlichen therapeutischen Optionen je nach KS-Variante und ihrer klinischen Präsentation bewusst zu sein.
Kutanes Angiosarkom
Angiosarkome machen zwischen 1 % und 2 % aller Sarkome aus, aber mindestens die Hälfte sind kutane Sarkome.25,26 Von den kutanen Sarkomen ist das Angiosarkom das vierthäufigste, nach KS, Dermatofibrosarkom und Leiomyosarkom. Das kutane Angiosarkom ist eines der kutanen Neoplasien mit der schlechtesten Prognose, mit einer 5-Jahres-Überlebensrate zwischen 10 % in den ältesten Serien27 und 30-50 % in den neuesten Serien.25,28,29 Es gibt 3 Hauptvarianten von kutanen Angiosarkomen: idiopathische Läsionen im Gesicht und auf der Kopfhaut älterer Patienten (Wilson-Jones-Angiosarkom), eine Variante, die etwa 50 % der kutanen Angiosarkome ausmacht, und 2 Formen sekundärer Angiosarkome, von denen eine in Bereichen chronischer Lymphödeme lokalisiert ist, insbesondere in den Armen von Frauen, die sich einer radikalen Mastektomie unterziehen (Stewart-Treves-Syndrom), und eine andere, die sich über Bereichen bestrahlter Haut entwickelt, insbesondere im Brustbereich von Frauen, die sich einer Strahlentherapie nach Brustkrebs unterziehen (Abb. 5). Es handelt sich um ein sehr aggressives kutanes Sarkom mit häufigem Lokalrezidiv und schlechter Prognose.27,30 Die einzige potenziell kurative Therapie ist die Operation mit oder ohne Strahlentherapie.
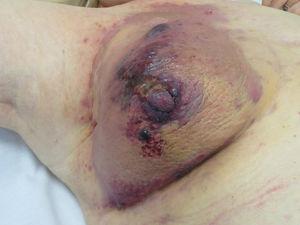
Angiosarkom. Rötlich-violette Papeln und Knötchen auf einer erythematös-violetten Plaque auf einer Brust, die zuvor wegen Krebs bestrahlt wurde.
Epidemiologie und klinische Diagnose
Die Inzidenz von kutanen Angiosarkomen ist schwer zu berechnen, aber Angiosarkome werden in den Vereinigten Staaten mit einer Rate von 0,4 Fällen pro Million Einwohner angegeben.31 Kutane Angiosarkome machen zwischen 35% und 60% aller Angiosarkome aus, mit einer ungefähren Inzidenz von 0,2 Fällen pro Million Einwohner. Sie überwiegen bei älteren Patienten mit einem Durchschnittsalter von 73 Jahren. Bei Kindern oder jungen Patienten sind diese Läsionen sehr selten. Das klassische Wilson-Jones-Angiosarkom überwiegt bei Männern und nach Strahlentherapie bei Frauen.29 Es betrifft auch überwiegend Weiße.25,29 Die Mehrzahl der idiopathischen Angiosarkome ist an Kopf und Hals lokalisiert (62 %), nach Bestrahlung überwiegen Läsionen am Rumpf (24 %), insbesondere im Brustbereich (nach Bestrahlung der Brust) und nach Lymphödemen an den Gliedmaßen (11 %). Die meisten Angiosarkome nach Bestrahlung treten nach einer Strahlentherapie aufgrund von Brustkrebs auf,32 aber es gibt auch Fälle in anderen bestrahlten Bereichen und nicht nur aufgrund maligner Prozesse. Die Latenzzeit zwischen der Strahlentherapie und der Entwicklung eines Angiosarkoms variiert, mit einem Mittelwert von 5 Jahren für Brustareale und 10 Jahren für andere Areale. Angiosarkome aufgrund eines Lymphödems treten vorwiegend in Bereichen mit chronischem Lymphödem an den Armen von Frauen auf, die sich einer radikalen Mastektomie unterzogen haben (Stewart-Treves-Syndrom),33 aber es wurden auch Fälle von Läsionen über Lymphödemen jeglicher Ätiologie berichtet. Die Zeit zwischen dem Auftreten eines Lymphödems und der Entwicklung eines Angiosarkoms variiert stark zwischen 1 Jahr und 30 Jahren.
Die anfänglich charakteristischste Form ist eine blaue, manchmal ödematöse Läsion mit einer schlecht definierten Häufigkeit, die anfangs eher unbemerkt bleibt, insbesondere auf der Kopfhaut von Patienten mit Haaren.34 Bei Angiosarkomen im Kopf- und Halsbereich kann das wahre Ausmaß der Läsion besser eingeschätzt werden, wenn der Patient seinen Kopf für einige Sekunden unter die Herzhöhe hält. Durch dieses Manöver wird der subklinische Anteil besser sichtbar, da er einen violetten Ton und ein ödematöses Aussehen annimmt.35 Später im klinischen Verlauf treten Papeln und Knötchen auf, gelegentlich mit Ulzeration und Blutung in fortgeschrittenen Phasen. In einigen Fällen treten Papeln und Knötchen direkt auf, ohne dass es vorher zu einer blutergussartigen Phase kommt. Die mittlere Größe bei Diagnosestellung beträgt 3-5 cm.28,29 Angiosarkome können als solitärer oder multifokaler Tumor auftreten, und die Läsion dehnt sich oft über die klinisch wahrnehmbaren Grenzen hinaus aus. Der klinische Verdacht auf ein kutanes Angiosarkom sollte durch eine Biopsie bestätigt werden.
Histologische Diagnose
Die 3 Typen von kutanen Angiosarkomen haben überlappende histopathologische Merkmale. Gut differenzierte Angiosarkome zeigen vaskuläre Lumina, die von abgeflachten Endothelzellen ausgekleidet sind, dissezierende Kollagenbündel, mit begrenzter Zellatypie. Die histologische Diagnose ist in diesen Phasen komplex und es ist nützlich, einige Endothelien mit prominenten, pleomorphen Zellen mit hyperchromatischen Kernen zu erkennen, die dazu neigen, hervorzustehen und einige Papillen zu bilden, mit mehreren Endothelzellschichten in den vaskulären Lumina.36,37 Die vaskulären Leitungen sind unregelmäßig und neigen dazu, anastomosierende Kanäle zu bilden (Abb. 6). In den am schlechtesten differenzierten Fällen sind die Tumorzellen epitheloid oder fusiform, mit ausgeprägter Atypie und reichlich Mitose und einem solideren Wachstumsmuster mit wenigen Gefäßlumina, so dass sie mit Karzinomen oder sogar Melanomen oder Fibrosarkomen verwechselt werden können. Das Vorhandensein von intrazytoplasmatischen Vakuolen als Ausdruck einer primitiven vaskulären Differenzierung kann in diesen Fällen sehr nützlich für den Verdacht auf eine korrekte Diagnose sein. Die Infiltration bei undifferenzierten Angiosarkomen ist sehr destruktiv und normale Bestandteile der Dermis und Hautanhangsgebilde verschwinden. Ein fleckiges entzündliches Infiltrat begleitet oft den Prozess und dieses Infiltrat kann manchmal so dicht sein, dass es einem Lymphom ähnelt.38 Das Ausmaß der begleitenden roten Blutkörperchen und des Hämosiderins ist sehr variabel. Unterschiedliche Differenzierungsgrade können oft in ein und demselben Angiosarkom zu finden sein. Die Epidermis kann normal, atrophisch oder ulzeriert sein. Das Ausmaß der Differenzierung kutaner Angiosarkome hat traditionell keinen Einfluss auf die Prognose, und daher wird der histologische Grad, anders als bei anderen Sarkomen, nicht für das Staging berücksichtigt.39 Dieser Ansatz könnte sich in Zukunft ändern, da eine aktuelle Studie mit der größten bisher veröffentlichten Serie von kutanen Angiosarkomen und Weichteilangiosarkomen mit 821 Patienten ein prognostisches Modell für das Staging entwickelt hat, das den histopathologischen Grad mit einbezieht.25
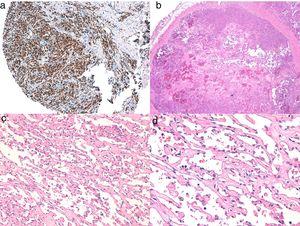
A, Immunhistochemie eines kutanen Angiosarkoms, das positiv für ERG ist (typischerweise mit einem Kernmuster). B, Angiosarkom mit Überwiegen von Bereichen mit vasoformativem Muster. C, D, Detailaufnahmen der neoplastischen Endothelzellen, die in diesem Fall prominent, aber ohne nennenswerte Atypien sind.
Die Untersuchung von Angiosarkomen sollte mit einem immunhistochemischen Panel vervollständigt werden, das ein Basis-Panel für Spindelzelltumore (CD31, Pancytokeratine, S110 und Aktin) und zusätzliche vaskuläre Marker (CD34, Erythroblasten-Transformations-spezifisches Gen , Podoplanin) umfasst. Einige Fälle von Angiosarkomen mit überwiegend epitheloiden Zellen können positiv für Zytokeratine sein; eine Positivität für vaskuläre Marker wie CD31, ERG und/oder Podoplanin kann jedoch ein undifferenziertes Karzinom ausschließen. In den letzten Jahren hat sich gezeigt, dass viele Angiosarkome eine MYC-Amplifikation/Überexpression aufweisen. In den meisten Studien wird eine MYC-Amplifikation in zwischen 50 % und 100 % der sekundären Angiosarkome gefunden, aber nicht generell in idiopathischen,40-42 aber eine MYC-Amplifikation oder Überexpression wurde auch in einigen idiopathischen Angiosarkomen nachgewiesen.43,44 Allerdings haben fast alle Studien das Fehlen einer MYC-Amplifikation oder Überexpression bei atypischen Gefäßproliferationen nach Bestrahlung gezeigt, so dass eine Positivität in einem zweifelhaften Fall von Gefäßproliferation im bestrahlten Bereich ein Angiosarkom fast ausschließt.
Der Ursprung des Angiosarkoms in Blut- oder Lymphgefäßen ist umstritten. Die Expression von CD31 oder CD34 ist in den am stärksten differenzierten Bereichen größer, aber in keinem der beiden Fälle ist sie konstant.45 Immunhistochemische Marker, die relativ spezifisch für Lymphgefäße sind, wie Podoplanin, D2-40, LYVE-1 und PROX-1, sind bei kutanen Angiosarkomen in der Regel positiv, oft mit einem gemischten immunhistochemischen Muster aus endothelialen Blutgefäßen und endothelialen Lymphgefäßen.38,46,47 Obwohl es sich um Ausnahmefälle handelt, wurden Fälle von kutanen Angiosarkomen berichtet, die S-100-Protein48 oder neuroendokrine Marker exprimieren.49
Staging
Obwohl es keine Leitlinien für das Management von kutanen Angiosarkomen gibt, wird angesichts der Tatsache, dass die häufigste Metastasierungsstelle die Lunge50 ist, gefolgt von den Lymphknoten,29,51 in der Regel eine thorakoabdominale Computertomographie nach der pathologischen Diagnose empfohlen. Diese bildgebende Untersuchung sollte bei einem Angiosarkom des Kopfes und Halses die Halsregion und bei abdominopelvinen Nachbestrahlungsangiosarkomen das Becken einschließen. Das Vorhandensein einer regionalen oder entfernten Dissemination ist bei Angiosarkomen nicht ungewöhnlich und wird in 30 % bzw. 10 % der Fälle berichtet.29
Es gibt kein spezifisches TNM-Staging für Angiosarkome, daher wird das TNM-Staging für Weichteilsarkome der American Joint Committee on Cancer-Klassifikation, angepasst an Angiosarkome, verwendet. Da der histologische Grad bei Angiosarkomen derzeit keinen Einfluss auf die Prognose hat, werden die Stadien IA undIIA in einigen Studien mit den Stadien IB undIIB zusammengefasst, die bei anderen Sarkomen nur durch den histologischen Grad unterschieden werden.
Behandlung
Die einzige Behandlung, die sich bei kutanen Angiosarkomen als potenziell kurativ erwiesen hat, ist die Operation, obwohl Heilung nur sporadisch auftritt. In inoperablen oder metastasierten Fällen hat jedoch die Strahlen- und/oder Chemotherapie eine anerkannte palliative Rolle. Darüber hinaus haben einige neuere Studien auch die Strahlentherapie als Adjuvans zur Operation in lokalisierten Fällen von Angiosarkomen einbezogen,50,51 und einige Autoren haben sogar die Bestrahlung regionaler Lymphknoten empfohlen,52 was aber nicht gängige Praxis ist. Das Problem bei der Operation von kutanen Angiosarkomen ist mitunter der multizentrische Charakter und die schlechte klinische Abgrenzung in anderen Fällen sowie die Tatsache, dass sie oft erst diagnostiziert werden, wenn die Läsionen größer als 5 cm sind. Zusätzlich zu diesen Faktoren sind die Patienten oft älter, was es schwieriger macht, geeignete chirurgische Ränder zu erhalten. Wenn die Tumoreigenschaften und der Allgemeinzustand des Patienten es zulassen, besteht die Behandlung des kutanen Angiosarkoms im Allgemeinen in der chirurgischen Exzision mit ausreichenden Rändern. Am meisten akzeptiert ist die chirurgische Entfernung mit einem 3 cm breiten Rand in Bezug auf die klinisch wahrnehmbaren Grenzen.53 Die Tiefe des Randes ist nicht gut festgelegt, da es sich um ein dermales Sarkom handelt, obwohl es sinnvoll erscheint, die Faszien zu erreichen, ohne sie zu exzidieren. In den am stärksten infiltrierenden Fällen sollte jedoch der Muskel mit einbezogen werden, um klare Ränder zu erreichen. Ränder mit Angiosarkom-Beteiligung haben sich in mehreren Studien als Faktor für eine schlechte Prognose erwiesen.28,30,51 Im Falle einer Brustbeteiligung schlagen die meisten Studien eine totale Mastektomie oder mehr oder weniger umfangreiche Exzisionen der bestrahlten Haut vor. In komplexen Fällen kann eine vorherige Biopsiekartierung bei der präoperativen Planung helfen. Wann immer es möglich ist, werden in der kutanen Onkologie direkte Verschlüsse, Transplantate oder Zweitverschlüsse bevorzugt, um die Nachsorge zu erleichtern und ein mögliches lokales Rezidiv nicht durch eine chirurgische Rekonstruktion zu verdecken, aber dies kann nach radikaleren Exzisionen von Angiosarkomen der Brust, die eine totale Mastektomie mit Bestrahlung der gesamten Haut erfordern, schwierig oder unmöglich sein. Bei Angiosarkomen, die durch ein Lymphödem verursacht werden, hat eine Studie die Berichte von 160 Patienten mit Stewart-Treves-Syndrom überprüft und festgestellt, dass eine Amputation im Vergleich zu einer radikalen Operation (mit einem Rand von 2 oder 3 cm) in diesen Fällen keinen Vorteil bringt, so dass eine Gliedmaßenamputation bei solchen Angiosarkomen nicht gerechtfertigt zu sein scheint.54
In Fällen, in denen eine Operation nicht möglich ist, d. h. bei multizentrischen oder ausgedehnten Läsionen oder bei solchen, die Bereiche betreffen, die eine Operation erschweren, ist die Strahlentherapie die Behandlung der Wahl.55 Die Dosis der Strahlentherapie für kutane Angiosarkome beträgt in der Regel 60 Gy, verteilt auf 20 Sitzungen zu je 3 Gy. Wenn sie adjuvant zur Operation eingesetzt wird, sind die Dosen ähnlich, es sei denn, sie ist für Angiosarkome nach der Bestrahlung indiziert, bei denen die Dosis in der Regel niedriger ist (45-50 Gy).
Die einzige Rolle der Chemotherapie beim Angiosarkom ist eine palliative Behandlung, die für rezidivierte oder metastasierte Läsionen reserviert ist, die einer Operation nicht zugänglich sind. Kürzlich wurde auch eine neoadjuvante Rolle für die Chemotherapie vor der Operation bei periorbitalen Läsionen vorgeschlagen.56 Die am häufigsten verwendeten Chemotherapien beim Angiosarkom sind Docetaxel,57,58 Paclitaxel,59 und liposomales Doxorrubicin,60 aber die aktuellen NCCN-Leitlinien schließen auch Vinorelbin, Sorafenib, Sunitinib und Bevacizumab ein, obwohl die Ergebnisse mit den drei letztgenannten antiangiogenen Wirkstoffen enttäuschend waren. Die Kombination mit Betablockern in dieser Phase der palliativen Behandlung kann für den Patienten von einigem Nutzen sein.61,62
Follow-up
Es gibt keine Standardrichtlinien für die Nachsorge von kutanen Angiosarkomen. In unserer Gruppe haben wir eine enge klinische Nachsorge, mit Kontrolluntersuchungen alle 3-6 Monate für die ersten paar Jahre und dann jährliche Kontrolluntersuchungen für 10 Jahre. Bei diesen Besuchen untersuchen wir die gesamte Haut und palpieren die entsprechenden territorialen Lymphknoten. Mindestens einmal im Jahr führen wir eine Laboranalyse und eine thorakoabdominopelvine Computertomographie-Studie durch.
Interessenkonflikte
Die Autoren erklären, dass sie keine Interessenkonflikte haben.