Der Sehnerv besteht aus etwa 1,2 Millionen Axonen, die ihren Ursprung in der Ganglienzellschicht der Netzhaut haben. Die Axone des Sehnervs sind stark von Oligodendrozyten myelinisiert, und die Axone regenerieren sich nicht, wenn sie verletzt werden. Daher verhält sich der Sehnerv eher wie ein Trakt der weißen Substanz als ein echter peripherer Nerv.
Der Sehnerv ist in die folgenden 4 Teile unterteilt:
-
Intraokulärer Teil (1 mm) (Sehnervenkopf)
-
Intraorbitaler Teil (25 mm)
-
Intrakanalikulärer Teil (5-9 mm)
-
Intrakranieller Teil (10-16 mm)
Der durchschnittliche Sehnervenkopf ist 1 mm tief, 1.5 mm breit, auf Höhe der Netzhaut 1,8 mm hoch und nach hinten hin etwas breiter. Der Sehnervenkopf sitzt an einem wichtigen Übergang von einem Bereich mit hohem Druck zu einem Bereich mit niedrigem Druck (intrakranieller Druck) und besteht aus 4 Zelltypen: Ganglienzellaxone, Astrozyten, kapillarassoziierte Zellen und Fibroblasten.
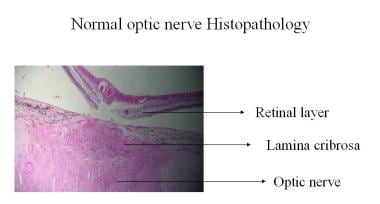 Normale Sehnervenhistopathologie.
Normale Sehnervenhistopathologie. Klinisch gesehen wird das vom Ophthalmoskop einfallende Licht durch die axonalen Fasern total reflektiert, und die anschließende Reflexion an den Kapillaren auf der Papillenoberfläche führt zu der charakteristischen gelb-rosa Farbe eines gesunden Sehnervenkopfes. Degenerierte Axone verlieren diese optische Eigenschaft, was die Blässe bei Optikusatrophie erklärt.
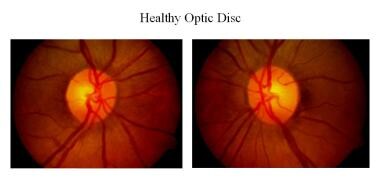 Gesunder Sehnervenkopf.
Gesunder Sehnervenkopf. Die Blutversorgung am Sehnervenkopf erfolgt durch piale Kapillaren, die aus dem Zinn-Haller-Kreis entspringen. Diese Kapillaren weisen eine Autoregulation auf und sind nicht undicht. Alternativ führt der Verlust dieser Kapillaren zu einer blass erscheinenden Papille. Der Kestenbaum-Kapillarzahl-Index ist die Anzahl der am Sehnervenkopf beobachteten Kapillaren. Der normale Wert liegt bei etwa 10. Bei einer Optikusatrophie reduziert sich die Anzahl dieser Kapillaren auf weniger als 6, während mehr als 12 auf einen hyperämischen Discus hinweisen.
Zu den histopathologischen Veränderungen, die bei einer Optikusatrophie beobachtet werden, gehören die folgenden:
-
Schrumpfung oder Verlust von Myelin und Achsenzylindern
-
Gliose
-
Vertiefung der physiologischen Exkavation mit Versteifung der Lamina cribrosa
-
Verbreiterung des Subarachnoidalraums mit redundanter Dura
-
Verbreiterung der pialen Septen
-
Durchtrennter Nerv führt zu knolligen axonalen Schwellungen (Cajal-Endbulben); können am vorderen Schnittende der Fasern beobachtet werden
Klassifikation
Optische Atrophie wird als pathologisch, ophthalmoskopisch oder ätiologisch klassifiziert.
Pathologische Optikusatrophie
Anterograde Degeneration (Wallersche Degeneration)
Die Degeneration beginnt in der Netzhaut und schreitet in Richtung des lateralen Genicularkörpers fort (z. B. toxische Retinopathie, chronisch einfaches Glaukom). Größere Axone zerfallen schneller als kleinere Axone.
Retrograde Degeneration
Die Degeneration beginnt im proximalen Teil des Axons und schreitet in Richtung Sehnervenkopf fort (z. B. Sehnervenkompression durch intrakraniellen Tumor).
Trans-synaptische Degeneration
Bei der trans-synaptischen Degeneration degeneriert ein Neuron auf der einen Seite einer Synapse als Folge des Verlustes eines Neurons auf der anderen Seite (z.B. bei Personen mit okzipitaler Schädigung, die entweder in utero oder im frühen Kindesalter entstanden ist).
Ophthalmoskopische Optikusatrophie
Primäre Optikusatrophie
Bei der primären Optikusatrophie (z. B. Hypophysentumor, Sehnerventumor, traumatische Optikusneuropathie, Multiple Sklerose) degenerieren die Sehnervenfasern in geordneter Weise und werden durch Säulen von Gliazellen ersetzt, ohne dass die Architektur des Sehnervenkopfes verändert wird. Die Papille ist kreideweiß und scharf abgegrenzt, die Netzhautgefäße sind normal. Die Lamina cribrosa ist gut definiert.
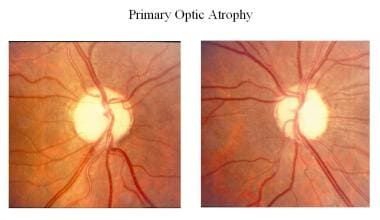 Primäre Optikusatrophie.
Primäre Optikusatrophie. Sekundäre Optikusatrophie
Bei Erkrankungen mit sekundärer Optikusatrophie (z. B. Papillenödem, Papillitis) ist die Atrophie sekundär zu einem Papillenödem (wie im Bild unten gezeigt). Die Sehnervenfasern zeigen eine ausgeprägte Degeneration mit einer übermäßigen Proliferation von Gliagewebe. Die Architektur ist verloren, was zu undeutlichen Rändern führt. Der Sehnervenkopf ist grau oder schmutzig grau, die Ränder sind schlecht definiert, und die Lamina cribrosa ist durch proliferierendes Fibrogliagewebe verdeckt. Es können hyaline Körper (Corpora amylacea) oder Drusen beobachtet werden. Peripapilläre Umhüllungen von Arterien sowie gewundene Venen können beobachtet werden. Auf den Gesichtsfeldern kann eine progressive Kontraktion der Gesichtsfelder beobachtet werden.
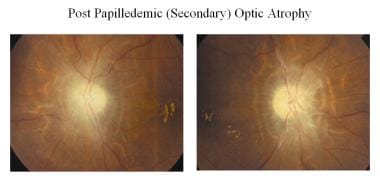 Optikusatrophie nach Papillenödem (sekundär).
Optikusatrophie nach Papillenödem (sekundär). Optische Atrophie nach Papillenödem (sekundär)
Optische Atrophie nach Papillenödem (sekundär) ist eine aufsteigende Form der Atrophie (z.B. Chorioretinitis, pigmentäre Netzhautdystrophie, zerebromakuläre Degeneration), die in der Regel durch Erkrankungen der Aderhaut oder der Netzhaut entsteht. Die Papille ist wächsern blass mit normalem Papillenrand, deutlicher Abschwächung der Arterien und normaler physiologischer Exkavation.
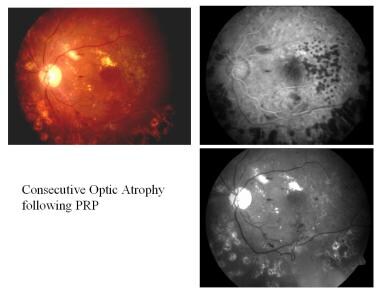 Konsekutive Optikusatrophie nach panretinaler Photokoagulation (PRP).
Konsekutive Optikusatrophie nach panretinaler Photokoagulation (PRP). Glaukomatöse Optikusatrophie
Auch bekannt als kavernöse Optikusatrophie, wird bei glaukomatöser Optikusatrophie eine ausgeprägte Exkavation der Papille beobachtet. Zu den Merkmalen gehören die vertikale Vergrößerung der Exkavation, die Sichtbarkeit der Laminarporen (Laminarpunktzeichen), die Rückwärtsverbiegung der Lamina cribrosa, die Bajonettierung und nasale Verschiebung der Netzhautgefäße sowie peripapilläre Halo und Atrophie. Splitterblutungen am Papillenrand können beobachtet werden.
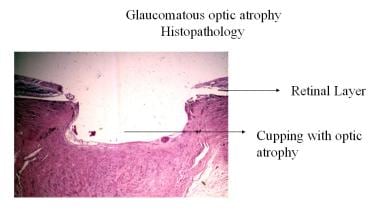 Glaukomatöse Optikusatrophie Histopathologie.
Glaukomatöse Optikusatrophie Histopathologie. Temporale Blässe
Temporale Blässe kann bei traumatischer oder ernährungsbedingter Optikusneuropathie beobachtet werden und ist am häufigsten bei Patienten mit Multipler Sklerose zu sehen, insbesondere bei solchen mit einer Vorgeschichte von Optikusneuritis. Die Papille ist blass mit einem klaren, abgegrenzten Rand und normalen Gefäßen, und die physiologische Blässe ist zeitlich deutlicher.
Etiologische Optikusatrophie
Hereditäre Atrophie
Diese wird unterteilt in kongenitale oder infantile Optikusatrophie (rezessive oder dominante Form), hereditäre Optikusatrophie nach Behr (autosomal rezessiv) und Lebersche Optikusatrophie. Mehrere hereditäre Optikusneuropathien, darunter die Optikusatrophie Typ 1 und die Lebersche Optikusatrophie, werden auf eine mitochondriale Dysfunktion in retinalen Ganglienzellen zurückgeführt.
Die autosomal-dominante Optikusatrophie Typ 1 wird durch Mutationen im OPA1-Gen auf Chromosom 3q29 verursacht. Das gebildete OPA1-Protein spielt eine Schlüsselrolle in einem Prozess, der als oxidative Phosphorylierung bezeichnet wird, und bei der Selbstzerstörung von Zellen (Apoptose). OPA1 ist ein integrales Pro-Fusionsprotein innerhalb der inneren Mitochondrienmembran. Mutationen im OPA1-Gen führen zu Sehproblemen bei Menschen mit einem Zusammenbruch der Strukturen, die visuelle Informationen von den Augen zum Gehirn übertragen. Bei den Betroffenen kommt es zunächst zu einem fortschreitenden Verlust von Nervenzellen innerhalb der Netzhaut, den sogenannten retinalen Ganglienzellen. Auf den Verlust dieser Zellen folgt die Degeneration (Atrophie) des Sehnervs.
Die X-chromosomale Optikusatrophie Typ 2 wird durch eine Mutation im OPA2-Gen mit der zytogenetischen Lokalisation Xp11.4-p11.21 verursacht. Der Patient präsentiert sich mit einem früh einsetzenden Sehverlust im Kindesalter mit langsamer Progression des Verlustes.
Hereditäre Optikusatrophie Typ 3 wird durch eine Mutation im OPA3-Gen mit zytogenetischer Lokalisation 19q13.32 verursacht. Die Mutation in diesem Gen ist mit einem im Kindesalter auftretenden Sehverlust mit Katarakt assoziiert. Sie kann auch mit einer Typ-III-Methylglutaconsäureurie assoziiert sein.
Leber hereditäre Optikusneuropathie entsteht durch mitochondriale Punktmutationen in der mtDNA 11778G>A, 14484T>C, oder 3460G>A Mutationen.
Die konsekutive Atrophie
Die konsekutive Atrophie ist eine aufsteigende Form der Atrophie (z. B. Chorioretinitis, pigmentäre Netzhautdystrophie, zerebromakuläre Degeneration), die meist auf Erkrankungen der Aderhaut oder der Netzhaut folgt.
Die zirkulatorische Atrophie (vaskulär)
Die zirkulatorische Atrophie ist eine ischämische Optikusneuropathie, die beobachtet wird, wenn der Perfusionsdruck des Ziliarkörpers unter den intraokularen Druck fällt. Zirkulatorische Atrophie wird bei Zentralarterienverschluss, Karotisarterienverschluss und kranialer Arteriitis beobachtet.
Metabolische Atrophie
Sie wird bei Erkrankungen wie Schilddrüsen-Ophthalmopathie, juvenilem Diabetes mellitus, ernährungsbedingter Amblyopie, toxischer Amblyopie, Tabak, Methylalkohol und Medikamenten (z. B. Ethambutol, Sulfonamiden) beobachtet.
Demyelinisierende Atrophie
Sie wird bei Erkrankungen wie Multipler Sklerose und Morbus Devic beobachtet.
Druck- oder Traktionsatrophie
Sie wird bei Erkrankungen wie Glaukom und Papillenödem beobachtet.
Postinflammatorische Atrophie
Sie wird bei Erkrankungen wie Sehnervenentzündung, Perineuritis sekundär zu Entzündungen der Hirnhäute und Sinus- und Orbitazellulitis beobachtet.
Traumatische Optikusneuropathie
Die genaue Pathophysiologie der traumatischen Optikusneuropathie ist nur unzureichend geklärt, obwohl die Abtrennung und Durchtrennung des Sehnervs, das Hämatom der Sehnervenscheide und die Einklemmung des Sehnervs durch einen eindringenden Fremdkörper oder ein knöchernes Fragment alle traumatische Formen der Sehnervenfehlfunktion widerspiegeln, die zu einer Optikusatrophie führen können.
Unabhängig von der Ätiologie ist die Optikusatrophie mit unterschiedlichen Graden der Sehstörung verbunden, die durch einen oder alle Sehnervenfunktionstests nachgewiesen werden können (siehe Andere Tests).
Die Strahlenoptische Neuropathie
Die Strahlenoptische Neuropathie tritt häufiger bei Strahlendosen von mindestens 5.000 Centigray auf. Sie kann die Folge einer Strahlenschädigung des Sehnervengefäßes oder des Sehnervenparenchyms selbst sein.