Sarcome de Kaposi
Le sarcome de Kaposi (KS) est une tumeur angioproliférative associée au virus de l’herpèsvirus humain de type 8 (HHV-8).1,2 Quatre variantes ont été décrites (tableau 1).
Epidémiologie et caractéristiques cliniques des différents types de sarcome de Kaposi.
| . | Sarcome de Kaposi classique | Sarcome de Kaposi endémique | Sarcome de Kaposi associé au VIH-.Sarcome de Kaposi associé au VIH | Sarcome de Kaposi associé à l’immunosuppression-Associated Kaposi Sarcoma |
|---|---|---|---|---|
| Age | >60 ans | 30-45 ans | 20-50 ans | 60 ans | Groupes à risque | L’ascendance méditerranéenne et juive | La population africaine | L’infection par le VIH et les HSH | Les receveurs de greffe, maladies auto-immunes | Site | Jambes | Jambes | Céphalique, oral, viscérale | Limbes | Manifestations extracutanées | Infréquentes | Souvent lymphoadénopathiques. | Fréquentes | Possibles |
| Evolution | Indolente | Progressive | Agressive, peut être retardée par un traitement antirétroviral | Variable, peut régresser avec une diminution de l’immunosuppression |
Abréviations : HSH, hommes ayant des rapports sexuels avec des hommes ; KS, sarcome de Kaposi.
Sarcome de Kaposi classique. Il s’agit d’une tumeur peu fréquente qui a tendance à toucher les hommes1,3 de la région méditerranéenne ou d’Europe centrale, avec une incidence comprise entre 0,18 et 13,2 cas/million.4 Elle survient plus fréquemment chez les hommes présentant un œdème chronique des jambes, un diabète sucré et des utilisateurs de corticostéroïdes. Les lésions se présentent sous forme de plaques ou de nodules érythémato-violacés uniques ou multiples à croissance lente, rarement associés à un lymphœdème des extrémités et à une atteinte gastro-intestinale et ganglionnaire. Son évolution clinique est indolente, et 2 % des patients meurent d’une maladie disséminée.
Sarcome de Kaposi endémique. Cette variante a été signalée en Afrique équatoriale et touche les jeunes adultes et les individus prépubères. Chez les adultes, la variante suit une évolution indolente ou agressive, avec une atteinte des tissus sous-cutanés et osseux, tandis que chez les enfants, elle se manifeste sous une forme agressive avec une atteinte ganglionnaire généralisée, une atteinte des organes internes et l’absence de lésions cutanées (ou des lésions limitées).5
Sarcome de Kaposi associé à l’immunosuppression (variante iatrogène). Cette variante a été rapportée chez des patients sous traitement par immunosuppresseurs et notamment chez des transplantés. Le risque de développer le KS est estimé entre 150 et 200 fois plus élevé que dans la population générale, avec un délai moyen d’apparition de 18mois.6
Sarcome de Kaposi associé au VIH (variante épidémique). Cette variante a été signalée chez des hommes infectés par le VIH ayant des rapports sexuels avec des hommes (HSH-VIH+). Avant l’ère de la thérapie antirétrovirale hautement active (HAART), on calculait que 25 % des HSH-VIH+ développeraient un sarcome de Kaposi, bien que ce pourcentage ait progressivement diminué2,7 (figures 1 et 2). Ces patients peuvent présenter une atteinte de la peau et des muqueuses, des ganglions lymphatiques, du tractus gastro-intestinal, des poumons, de la rate et du foie.7 Le KS chez les HSH-VIH- a également été rapporté, avec dans ce cas une évolution indolente.8
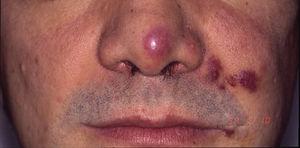
Sarcome de Kaposi chez un patient atteint du syndrome d’immunodéficience acquise. Plaques érythémato-violacées sur le bout du nez, le coin de la bouche et la joue gauche.
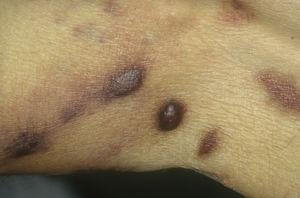
Sarcome de Kaposi classique sur les jambes. Plaques brunâtres érythémateuses sur la jambe.
Diagnostic
Le diagnostic est réalisé cliniquement, mais il est recommandé d’obtenir une confirmation par biopsie. L’étude histologique révèle des cellules fusiformes, une prolifération de vaisseaux irréguliers avec des formes en fente, une extravasation de globules rouges et un infiltrat leucocytaire avec des plasmocytes et des globules hyalins intra- et extracellulaires dans toute l’épaisseur du derme, ainsi que le signe dit du promontoire (figure 3). La réaction en chaîne par polymérase et la coloration immunohistochimique pour l’antigène nucléaire associé à la latence (LANA-1) du virus HHV-8 sont positives.1
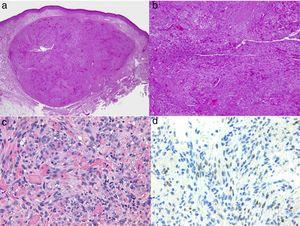
Histologie du sarcome de Kaposi.
A, Vue à faible grossissement du sarcome de Kaposi en phase nodulaire. Nodule dermique bien défini.
B, Lésion très cellulaire avec quelques lacunes sous forme de fissures.
C, Détail des cellules fusiformes et des globules rouges au sein des petits vaisseaux.
D, positivité nucléaire immunohistochimique pour le HHV-8 spécifique du sarcome de Kaposi.
Stadification et examens complémentaires
Sarcome de Kaposi classique. Compte tenu de la présentation clinique (âge, atteinte locale, atteinte peu fréquente des organes internes et évolution indolente), l’examen de la peau et des ganglions lymphatiques est suffisant. Des examens complémentaires sont réalisés si le patient présente des symptômes d’atteinte viscérale.5
Sarcome de Kaposi associé à l’immunosuppression. Comme pour le KS associé au VIH discuté ci-dessous, il n’existe pas de stadification consensuelle pour le KS associé à l’immunosuppression. Compte tenu de l’absence de consensus, les tests et les critères applicables recommandés sont généralement les mêmes que pour le KS associé au VIH.
Sarcome de Kaposi associé au VIH Il n’existe pas de système de stadification accepté pour cette variante. Il est recommandé d’effectuer une radiographie du thorax et, en cas d’anomalies évoquant une atteinte respiratoire, de réaliser une bronchoscopie ou une tomographie thoracique informatisée. Il est également recommandé d’écarter la présence de sang occulte fécal ; si ce test est positif, une endoscopie digestive doit être réalisée.
En 1989, le comité d’oncologie du groupe d’essais cliniques sur le sida a proposé une stadification (tableau 2) basée sur l’extension de la maladie, le nombre de cellules CD4T et l’atteinte systémique (infections opportunistes, symptômes B tels que fièvre, perte de poids ou diarrhée persistante, et état de performance de Karnofsky inférieur à 70 points). Une analyse prospective a montré que ces variables étaient liées à la survie des patients, les facteurs associés à un bon pronostic étant une maladie limitée, un nombre de lymphocytes CD4 supérieur à 150 cellules/mm3 et l’absence de compromission systémique9.
Stadification proposée par le comité d’oncologie des groupes d’essais cliniques sur le sida.
| Bon pronostic (0) | Petit pronostic (1) | |
|---|---|---|
| Tumeur | Tumeur uniquement présente dans la peau ou les ganglions lymphatiques, ou une atteinte buccale minime | Tumeur provoquant une ulcération ou une ulcération Sévère atteinte buccale Lésions dans le tractus gastro-intestinal Lésions sur les organes internes autres que les ganglions lymphatiques |
| Immunité | CD4 ≥ 200/μL | CD4… 200/μL | Aucune infection opportuniste ou muguet Aucun symptôme B État de performance de Karnofsky ≥ 70 |
Infections opportunistes ou muguet Symptômes B État de performance de Karnofsky 70 Autres maladies associées au VIH-.associées au VIH (par exemple, maladie neurologique ou lymphome) |
Abréviation ; KS, sarcome de Kaposi.
Fièvre prolongée, sueurs nocturnes, perte de poids de plus de 10% ou diarrhée durant plus de 15 jours.
Ces propositions de stadification ont été avancées avant que la HAART ne soit disponible et n’incluent pas la charge virale, de sorte que l’application de ce système de stadification a été limitée aux essais cliniques.
TraitementSarcome de Kaposi classique
Il existe peu d’essais cliniques comparatifs des différents traitements du KS classique. Les mêmes médicaments et régimes que ceux appliqués pour le KS épidémique sont généralement utilisés (tableau 3).
-
Lésions solitaires.
- –
Observation clinique. Compte tenu de l’âge des patients et de la faible mortalité, le suivi sans traitement peut être une option. En cas de lymphœdème, une compression élastique est recommandée.10
- –
Radiothérapie locale. La radiothérapie à basse énergie (100kV : 8Gy en une seule application ou 20-30Gy en dose fractionnée) est efficace pour les lésions isolées.10 Dans une analyse rétrospective de 68 patients traités par radiothérapie, une bonne réponse a été observée chez 85% des patients, avec une réponse complète chez 58% et une amélioration des symptômes chez 95%.3
- –
Excision chirurgicale. Dans le cas de lésions qui provoquent une gêne, comme celles des zones acrales, la chirurgie est recommandée. Dans une étude portant sur des lésions excisées chirurgicalement, on a constaté que les patients ne présentaient pas de rechute pendant une période moyenne de 15 mois après le traitement.3
- –
Cryothérapie. La cryothérapie peut être utilisée dans les lésions solitaires mesurant ≤1cm, notamment dans les régions acrales, avec des applications d’une durée de 30 à 60 secondes. La chirurgie et la cryothérapie ont toutes deux l’avantage de pouvoir être répétées avec de bons résultats.
- –
Traitement intralésionnel. Le traitement intralésionnel à la vinblastine (0,2mg/mL toutes les 2 semaines), à la vincristine (0,03-0,08mg) ou à l’interféron alfa (3-5MIU/3 fois par semaine pendant 4-5 semaines),10 l’application topique d’alitrétinoïne en gel à 0,1% (lésions maculaires) ou d’imiquimod en crème à 5% (3 fois par semaine pendant 24 semaines)10 sont des traitements recommandés dans la littérature, bien que l’expérience de ces régimes soit limitée5.
-
Maladie disséminée
- –
Doxorubicine liposomale pégylée (PLD) (20mg/m2 toutes les 3semaines). La PLD est le traitement de choix, sauf chez les patients souffrant de maladies cardiaques. La réponse est bonne ou excellente, avec des réponses partielles (diminution de 50% du nombre de lésions tumorales) ou une réponse complète durant 25 mois chez plus de 70% des patients.11 La durée de la chimiothérapie n’est pas bien établie, bien qu’il soit recommandé de maintenir le traitement pendant 1 à 2 cycles après une réponse clinique. Le traitement par PLD est généralement bien toléré, avec des effets secondaires limités, et il est moins cardiotoxique que le traitement par des composés traditionnels. Des doses cumulatives plus élevées et des traitements plus longs peuvent donc être administrés. Les toxicités les plus sévères (grade 3 et 4) sont peu fréquentes et incluent la neutropénie et l’anémie.12
- –
Vinblastine (3mg/m2/semaine/intraveineuse ou 6mg/m2/2 semaines/iv). La vinblastine offre de bons résultats, avec des taux de réponses entre 50 % et 90 %, bien qu’une leucopénie puisse survenir3,13.
- –
Les autres chimiothérapies associées à des taux de réponse élevés, mais avec des effets indésirables comprennent le paclitaxel iv (100mg/semaine), la bléomycine (15U/semaine pendant3semaines puis toutes les 3 semaines/intramusculaire), et l’étoposide oral (100mg/jour, 3-5jours par semaine).10 Il n’y a qu’un seul essai clinique randomisé qui a comparé la vinblastine iv à l’étoposide oral ; aucune différence significative n’a été trouvée dans la réponse ou la survie, mais une proportion plus élevée d’effets indésirables a été rapportée avec le traitement à l’étoposide13.
Algorithme de traitement du sarcome de Kaposi.
| Variante | Traitement | ||
|---|---|---|---|
| KS classique | Traitement de choix | Autres alternatives | |
| I. Lésions solitaires ou isolées | a. Envisager uniquement l’observation clinique. b. Radiothérapie locale ou excision chirurgicale |
Thérapie intralésionnelle | |
| II. Maladie disséminée | Doxorubicine liposomale | Vinblastine, paclitaxel, étoposide | Sarcome de Kaposi associé au VIH | I. Lésions solitaires ou isolées | Initier une HAART avec ou sans thérapie locale (radiothérapie, excision chirurgicale ou cryothérapie) | HAART et thérapie intralésionnelle |
| II. Maladie disséminée | HAART et doxorubicine liposomale | HAART avec paclitaxel | |
| KS iatrogène (associé à une immunosuppression) | Suspendre ou réduire la dose de l’immunosuppresseur | Suivre le régime de traitement du KS associé au VIH-.associé au VIH | |
Abréviations : HAART, traitement antirétroviral hautement actif ; KS, sarcome de Kaposi
Sarcome de Kaposi associé au VIH
-
Lésions solitaires ou limitées
- –
HAART en monothérapie ou associé à un traitement local (Fig. 4). La HAART est administrée comme traitement initial, étant donné qu’il a été démontré que ces agents reconstituent la fonction immunitaire et diminuent l’incidence et la gravité du sarcome,2,7 avec réduction ou disparition des lésions. Dans les lésions symptomatiques et antiesthétiques, la chirurgie14, l’électrocoagulation ou la cryothérapie sont des options. La vinblastine intralésionnelle15 (0,2 à 0,3mg/mL, en administrant 0,1mL pour chaque lésion de 0,5cm2) ou la radiothérapie à basse énergie (100kV, doses comprises entre 8Gy/1fraction ou 30Gy/10fractions, réponse clinique complète à plus de 95%) peuvent être envisagées.16 La survie sans lésion à 5 ans après un traitement HAART, avec ou sans thérapie locale était de 92% dans une série de plus de 400 cas.14
 Figure 4.
Figure 4.Algorithme de traitement du sarcome de Kaposi associé au VIH.
Abréviations : HAART, thérapie antirétrovirale hautement active ; IRS, syndrome de reconstitution immunitaire.
Adapté du Groupe de consensus pour le traitement du sarcome de Kaposi associé au VIH. Réunion de consensus. Barcelone : Saned ; 1998.
(0,12MB).Maladie disséminée
Le traitement systémique est recommandé chez les patients traités par HAART et présentant une atteinte cutanée étendue (plus de 15 à 25 lésions), un gonflement intense, un KS cutané qui n’a pas répondu au traitement local ou qui progresse rapidement, un KS associé à un syndrome de reconstitution immunitaire ou une atteinte symptomatique des organes internes.
- –
PLD (20mg/m2 toutes les 3 semaines). La HAART et la PLD doivent être commencées en même temps,17 car l’association est plus efficace que la HAART seule.18 Plusieurs cures sont généralement administrées en fonction de la réponse clinique. Une réponse complète/partielle est obtenue avec la thérapie combinée dans 80 % des cas19, avec une survie à 5 ans supérieure à 85 %. Les rechutes sont limitées (13%) et surviennent au cours de la première année après la fin du traitement.20 La réponse à la PLD est plus élevée que l’association de bléomycine, vincristine ou vinblastine et doxorubicine non liposomale,21 et daunorubicine liposomale.22
- –
Paclitaxel (100mg/m2 toutes les 2 semaines). Le paclitaxel est un traitement de deuxième ligne avec des réponses favorables chez 71% des patients,23 mais avec des taux de survie inférieurs à ceux de la PLD et des taux plus élevés de toxicités de grade 3-5.14,24 Une prémédication avec la dexaméthasone est nécessaire et des interactions médicamenteuses graves peuvent se produire avec les agents antirétroviraux.
- –
Autres thérapies. D’autres médicaments, tels que l’étoposide, la vinorelbine, l’interleukine12, le bevacizumab et l’imatinib ont également été utilisés, mais l’expérience est limitée.5
Sarcome de Kaposi associé à une immunosuppression
- –
Suspendre l’immunosuppresseur ou réduire la dose. La suspension ou la réduction de la dose de l’immunosuppresseur peut induire des rémissions spontanées. Si cela est inefficace ou peu pratique, on applique généralement les approches utilisées dans le KS associé au VIH.5
Conclusions
Le KS est une tumeur angioproliférative dont les différents sous-types sont associés à l’âge avancé, à certaines populations africaines, à l’immunosuppression iatrogène ou à l’infection par le VIH. Bien que la multithérapie ait conduit à une diminution spectaculaire de l’incidence et de la gravité du KS chez les personnes infectées par le VIH, il est important de connaître les différentes options thérapeutiques en fonction de la variante du KS et de sa présentation clinique.
Angiosarcome cutané
Les angiosarcomes représentent entre 1 % et 2 % de tous les sarcomes, mais au moins la moitié sont cutanés.25,26 Parmi les sarcomes cutanés, l’angiosarcome est le quatrième plus fréquent, derrière le KS, le dermatofibrosarcome et le léiomyosarcome. L’angiosarcome cutané est l’un des néoplasmes cutanés de plus mauvais pronostic, avec une survie à 5 ans comprise entre 10% dans les séries les plus anciennes27 et 30%-50% dans les plus récentes25,28,29. Il existe 3 principales variantes d’angiosarcomes cutanés : des lésions idiopathiques sur le visage et le cuir chevelu des patients âgés (angiosarcome de Wilson-Jones), une variante qui représente environ 50 % des angiosarcomes cutanés, et 2 formes d’angiosarcomes secondaires, l’une localisée dans les zones de lymphoedème chronique, en particulier dans les bras des femmes qui subissent une mastectomie radicale (syndrome de Stewart-Treves) et l’autre qui se développe sur des zones de peau irradiée, en particulier dans la région pectorale des femmes qui subissent une radiothérapie après un cancer du sein (Fig. 5). Il s’agit d’un sarcome cutané très agressif avec de fréquentes récidives locales et un mauvais pronostic.27,30 La seule thérapie potentiellement curative est la chirurgie avec ou sans radiothérapie.
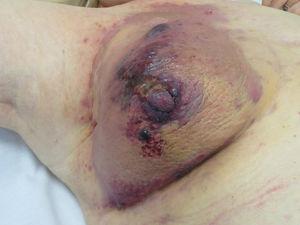 Figure 5.
Figure 5.Angiosarcome. Papules et nodules rougeâtres-violacés sur une plaque érythémato-violacée sur un sein préalablement irradié pour un cancer.
(0,07MB).Épidémiologie et diagnostic clinique
L’incidence des angiosarcomes cutanés est difficile à calculer, mais l’ensemble des angiosarcomes est rapporté à un taux de 0,4 cas par million d’habitants aux États-Unis31. Les angiosarcomes cutanés représentent entre 35 et 60 % de tous les angiosarcomes, avec une incidence approximative de 0,2 cas par million d’habitants. Ils prédominent chez les patients âgés, avec un âge moyen de 73 ans. Ces lésions sont très rares chez les enfants ou les jeunes patients. L’angiosarcome classique de Wilson-Jones prédomine chez les hommes et après radiothérapie chez les femmes.29 Il touche également majoritairement les blancs.25,29 La majorité des angiosarcomes idiopathiques sont localisés sur la tête et le cou (62%), les lésions post-radiation prédominent sur le tronc (24%), en particulier dans la région pectorale (post-radiation du sein) et après un lymphœdème sur les membres (11%). La plupart des angiosarcomes post-radiation apparaissent après une radiothérapie due à un cancer du sein32, mais il existe des cas dans d’autres zones irradiées et pas seulement en raison de processus malins. Le temps de latence entre la radiothérapie et le développement de l’angiosarcome est variable, avec une moyenne de 5 ans pour les sites mammaires et de 10 ans pour les autres sites. Les angiosarcomes dus au lymphoedème se produisent principalement dans les zones de lymphoedème chronique sur les bras des femmes qui subissent une mastectomie radicale (syndrome de Stewart-Treves),33 mais des cas de lésions sur un lymphoedème de toute étiologie ont été rapportés. Le délai entre la présentation du lymphœdème et le développement des angiosarcomes varie fortement entre 1 an et 30 ans.
La forme initiale la plus caractéristique est une lésion de type ecchymose, parfois œdémateuse, de fréquence mal définie, qui a tendance à passer inaperçue au début, notamment sur le cuir chevelu des patients ayant des cheveux.34 Dans le cas d’un angiosarcome de la tête et du cou, l’étendue réelle de la lésion peut être mieux appréciée si le patient tient sa tête sous le niveau du cœur pendant quelques secondes. Cette manœuvre rendra plus visible la partie subclinique qui prend un ton violacé et un aspect œdémateux.35 Plus tard dans l’évolution clinique, des papules et des nodules apparaissent, avec parfois des ulcérations et des saignements dans les phases avancées. Certains cas présentent directement des papules et des nodules sans pratiquement aucune phase préalable d’ecchymose. La taille moyenne au moment du diagnostic est de 3 à 5 cm.28,29 Les angiosarcomes peuvent se présenter comme une tumeur solitaire ou multifocale et la lésion s’étend souvent au-delà des limites cliniquement appréciables. La suspicion clinique d’angiosarcome cutané doit être confirmée par une biopsie.
Diagnostic histologique
Les 3 types d’angiosarcomes cutanés ont des caractéristiques histopathologiques qui se chevauchent. Les angiosarcomes bien différenciés présentent des luminescences vasculaires bordées de cellules endothéliales aplaties, des faisceaux de collagène disséquants, avec des atypies cellulaires limitées. Le diagnostic histologique est complexe dans ces phases et il est utile de reconnaître certains endothéliums avec des cellules proéminentes, pléomorphes avec des noyaux hyperchromatiques qui ont tendance à faire saillie et à créer quelques papilles, avec plusieurs couches de cellules endothéliales dans la lumière vasculaire.36,37 Les conduits vasculaires sont irréguliers et ont tendance à former des canaux anastomosés (Fig. 6). Dans les cas les moins bien différenciés, les cellules tumorales sont épithélioïdes ou fusiformes, avec des atypies marquées et des mitoses abondantes et un schéma de croissance plus solide avec peu de lumières vasculaires, de sorte qu’elles peuvent être confondues avec des carcinomes, voire des mélanomes ou des fibrosarcomes. La présence de vacuoles intracytoplasmiques comme expression d’une différenciation vasculaire primitive peut être très utile pour suspecter un diagnostic correct dans ces cas. L’infiltration dans le cas d’angiosarcomes indifférenciés est très destructive et les composants normaux du derme et des appendices cutanés disparaissent. Un infiltrat inflammatoire parcellaire accompagne souvent le processus et cet infiltrat peut parfois être si dense qu’il ressemble à un lymphome.38 L’étendue des globules rouges et de l’hémosidérine qui l’accompagnent est très variable. On peut souvent trouver différents degrés de différenciation dans un même angiosarcome. L’épiderme peut être normal, atrophique ou ulcéré. Le degré de différenciation des angiosarcomes cutanés n’est pas traditionnellement considéré comme ayant un impact sur le pronostic, et donc, contrairement à d’autres sarcomes, le grade histologique n’est pas pris en compte pour la stadification39. Cette approche pourrait changer à l’avenir, car une étude récente avec la plus grande série d’angiosarcomes cutanés et d’angiosarcomes des tissus mous publiée à ce jour, avec 821 patients, a développé un modèle pronostique pour la survie qui inclut le grade histopathologique.25
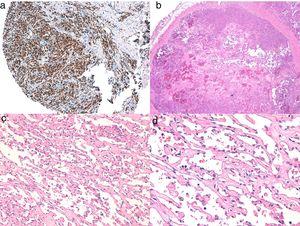 Figure 6.
Figure 6.A, Immunohistochimie d’un angiosarcome cutané qui est positif pour l’ERG (typiquement avec un motif nucléaire). B, Angiosarcome avec prédominance de zones à motif vasoformatif. C, D, Images détaillées des cellules endothéliales néoplasiques, qui dans ce cas sont proéminentes mais sans atypies notables.
(0.71MB).L’étude d’un angiosarcome doit être complétée par un panel immunohistochimique comprenant un panel de base pour les tumeurs à cellules fusiformes (CD31, pancytokératines, S110 et actine) et des marqueurs vasculaires supplémentaires (CD34, erythroblast transformation-specific-related gene , podoplanin). Certains cas d’angiosarcome avec prédominance de cellules épithélioïdes peuvent être positifs pour les cytokératines ; cependant, la positivité pour les marqueurs vasculaires tels que CD31, ERG, et/ou podoplanine peut exclure un carcinome indifférencié. Ces dernières années, il a été démontré que de nombreux angiosarcomes présentent une amplification/ surexpression de MYC. Dans la plupart des études, l’amplification de MYC est trouvée dans 50 à 100 % des angiosarcomes secondaires, mais généralement pas dans les angiosarcomes idiopathiques,40-42 mais l’amplification ou la surexpression de MYC a également été détectée dans certains angiosarcomes idiopathiques.43,44 Cependant, presque toutes les études ont montré l’absence d’amplification ou de surexpression de MYC dans les proliférations vasculaires atypiques post-radiation, et donc la positivité dans un cas douteux de prolifération vasculaire dans la zone irradiée exclut presque l’angiosarcome.
L’origine de l’angiosarcome dans les vaisseaux sanguins ou lymphatiques est sujette à débat. L’expression du CD31 ou du CD34 est plus importante dans les zones les plus différenciées, mais dans aucun des deux cas elle n’est constante.45 Les marqueurs immunohistochimiques relativement spécifiques des vaisseaux lymphatiques, tels que la podoplanine, D2-40, LYVE-1 et PROX-1, sont généralement positifs dans les angiosarcomes cutanés, souvent exprimés avec un schéma immunohistochimique mixte de vaisseaux sanguins endothéliaux et de vaisseaux lymphatiques endothéliaux38,46,47. Bien qu’exceptionnels, des cas ont été rapportés d’angiosarcomes cutanés exprimant la protéine S-100,48 ou des marqueurs neuroendocriniens.49
Stadification
Bien qu’il n’y ait pas de directives pour la gestion de l’angiosarcome cutané, étant donné que le site le plus fréquent de métastase est le poumon50 suivi des ganglions lymphatiques,29,51 la tomographie thoraco-abdominale est habituellement recommandée après le diagnostic pathologique. Cette étude d’imagerie doit inclure la région cervicale dans le cas d’un angiosarcome de la tête et du cou et le pelvis dans le cas d’angiosarcomes abdominopelviens post-radiation. La présence d’une dissémination régionale ou à distance n’est pas rare avec les angiosarcomes, et est rapportée dans 30 % et 10 % des cas, respectivement.29
Il n’existe pas de stadification TNM spécifique aux angiosarcomes, c’est pourquoi on utilise la stadification TNM pour les sarcomes des tissus mous de la classification de l’American Joint Committee on Cancer, adaptée aux angiosarcomes. Dans le cas des angiosarcomes, le grade histologique n’étant pas actuellement considéré comme ayant un impact sur le pronostic, les stades IA etIIA sont regroupés dans certaines études avec les stades IB etIIB, qui dans d’autres sarcomes sont différenciés uniquement par le grade histologique.
Traitement
Le seul traitement démontré comme potentiellement curatif dans les angiosarcomes cutanés est la chirurgie, bien que la guérison ne soit que sporadique. Cependant, dans les cas inopérables ou métastatiques, la radiothérapie et/ou la chimiothérapie ont un rôle palliatif reconnu. De plus, certaines études récentes ont également inclus la radiothérapie comme adjuvant à la chirurgie dans les cas localisés d’angiosarcome,50,51 et certains auteurs ont même recommandé d’irradier les ganglions lymphatiques régionaux,52 mais ce n’est pas une pratique courante. Le problème de la chirurgie dans les angiosarcomes cutanés est parfois le caractère multicentrique et la mauvaise délimitation clinique dans d’autres cas, ainsi que le fait qu’ils sont souvent diagnostiqués lorsque les lésions dépassent 5 cm de taille. En plus de ces facteurs, les patients sont souvent âgés, ce qui rend plus difficile l’obtention de marges chirurgicales appropriées. En général, si les caractéristiques de la tumeur et l’état général du patient le permettent, le traitement de l’angiosarcome cutané est l’excision chirurgicale avec des marges suffisantes. La plus acceptée est la chirurgie avec une marge de 3 cm par rapport aux limites cliniquement appréciables.53 La profondeur de la marge n’est pas bien établie, étant donné qu’il s’agit d’un sarcome dermique, bien qu’il semble raisonnable d’atteindre les fascias sans les exciser. Dans les cas les plus infiltrés, cependant, le muscle doit être inclus pour obtenir des marges nettes. Il a été démontré que les marges avec atteinte de l’angiosarcome sont un facteur de mauvais pronostic dans plusieurs études.28,30,51 En cas d’atteinte du sein, la plupart des études suggèrent une mastectomie totale ou des excisions plus ou moins étendues de la peau irradiée. Dans les cas complexes, une cartographie préalable par biopsie peut aider à la planification pré-chirurgicale. Dans la mesure du possible, en oncologie cutanée, on privilégie les fermetures directes, les greffes ou les fermetures de seconde intention pour faciliter le suivi et ne pas masquer une éventuelle récidive locale lors de la reconstruction chirurgicale, mais cela peut être difficile ou impossible après des excisions plus radicales d’angiosarcomes mammaires qui nécessitent une mastectomie totale avec irradiation de toute la peau. Dans le cas des angiosarcomes causés par un lymphœdème, une étude a examiné les rapports de 160 patients atteints du syndrome de Stewart-Treves et a constaté l’absence de bénéfice de l’amputation par rapport à la chirurgie radicale (avec une marge de 2 ou 3 cm) dans ces cas, et l’amputation des membres ne semble donc pas justifiée dans ces angiosarcomes54.
Dans les cas où la chirurgie est impossible, c’est-à-dire les lésions multicentriques ou étendues, ou celles qui affectent des zones compliquant la chirurgie, la radiothérapie est le traitement de choix.55 La dose de radiothérapie pour les angiosarcomes cutanés est habituellement de 60 Gy répartis en 20 séances de 3 Gy chacune. Lorsqu’elle est utilisée comme adjuvant à la chirurgie, les doses sont similaires, sauf lorsqu’elle est indiquée pour les angiosarcomes post-radiation, dans lesquels la dose sera généralement plus faible (45-50 Gy).
Le seul rôle de la chimiothérapie dans l’angiosarcome est un traitement palliatif, réservé aux lésions récidivantes ou métastatiques non accessibles à la chirurgie. Un rôle néoadjuvant a également été récemment proposé pour la chimiothérapie avant la chirurgie dans les sites périorbitaires.56 Les chimiothérapies les plus utilisées dans l’angiosarcome sont le docétaxel,57,58 le paclitaxel,59 et la doxorrubicine liposomale,60 mais les directives actuelles du NCCN incluent également la vinorelbine, le sorafénib, le sunitinib et le bevacizumab, bien que les résultats avec ces 3 derniers agents antiangiogéniques aient été décevants. L’association avec des bêtabloquants dans cette phase du traitement palliatif peut être d’un certain bénéfice pour le patient.61,62
Suivi
Il n’existe pas de directives standard pour le suivi des angiosarcomes cutanés. Dans notre groupe, nous avons un suivi clinique étroit, avec des contrôles tous les 3-6 mois pendant les premières années, puis des contrôles annuels pendant 10 ans. Lors de ces visites, nous examinons la peau entière et palpons les ganglions lymphatiques territoriaux correspondants. Au moins une fois par an, nous effectuons une analyse de laboratoire et une étude tomographique thoracoabdominopelvienne.
Conflits d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts.