Sarcoma di Kaposi
Il sarcoma di Kaposi (KS) è un tumore angioproliferativo associato al virus herpesvirus umano tipo 8 (HHV-8).1,2 Sono state descritte quattro varianti (Tabella 1).
Epidemiologia e caratteristiche cliniche dei diversi tipi di sarcoma di Kaposi.
| KS classico | KS endemico | HIV-associato al sarcoma di Kaposi | Immunosoppressione-Associated Kaposi Sarcoma | |
|---|---|---|---|---|
| Età | >60 anni | 30-45 anni | 20-50 anni | 60 anni |
| Gruppi a rischio | Discesa mediterranea ed ebraica | Popolazione africana | Infezione da HIV e MSM | Ricevente trapianti, malattie autoimmuni |
| Sito | Gambe | Gambe | Cephalic, orale, viscerale | Limbs |
| Extracutaneo manifestazioni | Infrequente | Spesso linfoadenopatico | Frequente | Possibile |
| Esito | Corso indolente | Progressivo | Aggressivo, può essere ritardato con la terapia antiretrovirale | Variabile, può regredire con la diminuzione dell’immunosoppressione |
Abbreviazioni: MSM, uomini che fanno sesso con uomini; KS, sarcoma di Kaposi.
Sarcoma di Kaposi classico. Si tratta di un tumore non comune che tende a colpire gli uomini1,3 nella regione mediterranea o dell’Europa centrale, con un’incidenza compresa tra 0,18 e 13,2 casi/milione.4 Si verifica più frequentemente negli uomini con edema cronico delle gambe, diabete mellito e consumatori di corticosteroidi. Le lesioni si presentano come placche o noduli eritemato-violacei singoli o multipli a crescita lenta, in rare occasioni associati a linfedema alle estremità e coinvolgimento gastrointestinale e linfonodale. Ha un decorso clinico indolente, e il 2% dei pazienti muore di malattia disseminata.
Sarcoma di Kaposi endemico. Questa variante è stata riportata nell’Africa equatoriale e colpisce giovani adulti e individui prepuberi. Negli adulti, la variante segue un decorso indolente o aggressivo, con coinvolgimento del tessuto sottocutaneo e osseo, mentre nei bambini si manifesta in una forma aggressiva con coinvolgimento linfonodale generalizzato, coinvolgimento degli organi interni e assenza (o limitazione) delle lesioni cutanee.5
Sarcoma di Kaposi associato all’immunosoppressione (variante iatrogena). Questa variante è stata segnalata in pazienti in trattamento con immunosoppressori e in particolare nei riceventi di trapianti. Si stima che il rischio di sviluppare il KS sia tra 150 e 200 volte maggiore che nella popolazione generale, con un tempo medio di insorgenza di 18 mesi.6
Sarcoma di Kaposi associato all’HIV (variante epidemica). Questa variante è stata riportata in uomini con infezione da HIV che fanno sesso con altri uomini (MSM-HIV+). Prima dell’era della terapia antiretrovirale altamente attiva (HAART), si calcolava che il 25% degli MSM-HIV+ avrebbe sviluppato il KS, sebbene questa percentuale sia diminuita progressivamente2,7 (Figg. 1 e 2). Questi pazienti possono avere un coinvolgimento della pelle e delle mucose, dei linfonodi, del tratto gastrointestinale, dei polmoni, della milza e del fegato.7 È stato riportato anche il KS in MSM-HIV-, in questo caso con un decorso indolente.8
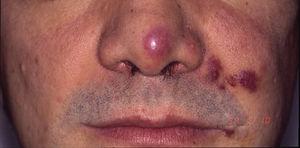 Sarcoma di Kaposi in un paziente con sindrome da immunodeficienza acquisita. Placche eritemato-violacee sulla punta del naso, sull’angolo della bocca e sulla guancia sinistra.
Sarcoma di Kaposi in un paziente con sindrome da immunodeficienza acquisita. Placche eritemato-violacee sulla punta del naso, sull’angolo della bocca e sulla guancia sinistra.
Sarcoma di Kaposi in un paziente con sindrome da immunodeficienza acquisita. Placche eritemato-violacee sulla punta del naso, sull’angolo della bocca e sulla guancia sinistra.
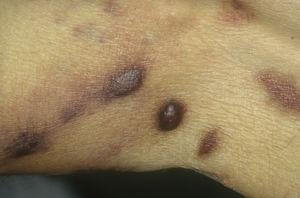
Sarcoma di Kaposi classico sulle gambe. Placche eritematose brunastre sulla gamba.
Diagnosi
La diagnosi viene eseguita clinicamente, ma si raccomanda di ottenere la conferma mediante biopsia. Lo studio istologico rivela cellule fusate, proliferazione di vasi irregolari con forme a fessura, stravaso di globuli rossi e infiltrato leucocitario con plasmacellule e globuli ialini intra ed extracellulari in tutto lo spessore del derma, così come il cosiddetto segno del promontorio (Fig. 3). La reazione a catena della polimerasi e la colorazione immunoistochimica per l’antigene nucleare associato alla latenza (LANA-1) del virus HHV-8 sono positive.1
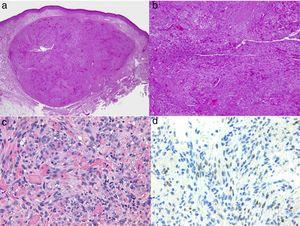 Estologia del sarcoma di Kaposi. A, vista a basso ingrandimento del sarcoma di Kaposi in fase nodulare. Nodulo dermico ben definito. B, lesione altamente cellulare con alcune lacune sotto forma di fessure. C, Dettaglio di cellule fusate e globuli rossi all’interno dei piccoli vasi. D, Positività nucleare immunoistochimica per HHV-8 specifica per il sarcoma di Kaposi.
Estologia del sarcoma di Kaposi. A, vista a basso ingrandimento del sarcoma di Kaposi in fase nodulare. Nodulo dermico ben definito. B, lesione altamente cellulare con alcune lacune sotto forma di fessure. C, Dettaglio di cellule fusate e globuli rossi all’interno dei piccoli vasi. D, Positività nucleare immunoistochimica per HHV-8 specifica per il sarcoma di Kaposi.
Estologia del sarcoma di Kaposi.
A, vista a basso ingrandimento del sarcoma di Kaposi in fase nodulare. Nodulo dermico ben definito.
B, lesione altamente cellulare con alcune lacune sotto forma di fessure.
C, dettaglio di cellule fusate e globuli rossi all’interno dei piccoli vasi.
D, Positività nucleare immunoistochimica per l’HHV-8 specifico del sarcoma di Kaposi.
Stadiazione e test complementari
Sarcoma di Kaposi classico. In considerazione della presentazione clinica (età, coinvolgimento locale, coinvolgimento infrequente degli organi interni e decorso indolente), l’esame della pelle e dei linfonodi è sufficiente. Test complementari vengono eseguiti se il paziente presenta sintomi di compromissione viscerale.5
Sarcoma di Kaposi associato all’immunosoppressione. Come per il KS associato all’HIV discusso più avanti, non c’è un consenso sulla stadiazione del KS associato all’immunosoppressione. Data la mancanza di consenso, i test e i criteri applicabili raccomandati sono solitamente gli stessi del KS associato all’HIV.
Sarcoma di Kaposi associato all’HIV Non esiste un sistema di stadiazione accettato per questa variante. Si raccomanda la radiografia del torace e, in caso di anomalie suggestive di compromissione respiratoria, si dovrebbe eseguire una broncoscopia o una tomografia computerizzata del torace. Si raccomanda anche di escludere la presenza di sangue occulto fecale; se questo test è positivo, deve essere eseguita un’endoscopia digestiva.
Nel 1989, il Comitato Oncologico dell’AIDS Clinical Trials Group ha proposto una stadiazione (Tabella 2) basata sull’estensione della malattia, sulla conta delle cellule CD4T e sulla compromissione sistemica (infezioni opportunistiche, sintomi B come febbre, perdita di peso o diarrea persistente, e performance status Karnofsky inferiore a 70 punti). Un’analisi prospettica ha mostrato che queste variabili erano correlate alla sopravvivenza dei pazienti, con fattori associati a una buona prognosi come una malattia limitata, una conta dei linfociti CD4 superiore a 150 cellule/mm3 e l’assenza di compromissione sistemica.9
Staging proposto dall’AIDS Clinical Trials Groups Oncology Committee.
| Prognosi buona (0) | Prognosi Pessima (1) | |
|---|---|---|
| Tumore | Tumore trovato solo nella pelle o nei linfonodi, o coinvolgimento orale minimo | Tumore che causa o ulcerazione Interessamento orale grave Lesioni diKS nel tratto gastrointestinale Lesioni diKS su organi interni diversi dai linfonodi |
| Immunità | CD4 ≥ 200/μL | CD4 200/μL |
| Malattie sistemiche | Nessuna infezione opportunistica o mughetto Nessun sintomo B Karnofsky performance status ≥ 70 |
Infezioni opportunistiche o mughetto B symptomsa Karnofsky performance status 70 Altro HIV-associate all’HIV (per es, malattia neurologica o linfoma) |
Abbreviazione; KS, sarcoma di Kaposi.
Febbre prolungata, sudorazione notturna, perdita di peso superiore al 10% o diarrea che dura più di 15 giorni.
Queste proposte di stadiazione sono state avanzate prima che la HAART fosse disponibile e non includono la carica virale, e quindi l’applicazione di questo sistema di stadiazione è stata limitata agli studi clinici.
TrattamentoSarcoma di Kaposi classico
Ci sono pochi studi clinici comparativi dei diversi trattamenti del KS classico. Di solito vengono utilizzati gli stessi farmaci e gli stessi regimi applicati per il KS epidemico (Tabella 3).
-
Le lesioni solitarie.
- –
Osservazione clinica. Data l’età dei pazienti e la bassa mortalità, il follow-up senza trattamento può essere un’opzione. In caso di linfedema, si raccomanda la compressione elastica.10
- –
Radioterapia locale. La radioterapia a bassa energia (100kV: 8Gy in una singola applicazione o 20-30Gy come dose frazionata) è efficace per lesioni isolate.10 In un’analisi retrospettiva di 68 pazienti trattati con radioterapia, è stata osservata una buona risposta nell’85% dei pazienti, con una risposta completa nel 58% e un miglioramento dei sintomi nel 95%.3
- –
Escissione chirurgica. Nel caso di lesioni che causano disagio, come quelle nelle aree acrali, si raccomanda la chirurgia. In uno studio sulle lesioni asportate chirurgicamente, i pazienti sono risultati liberi da ricadute per una media di 15 mesi dopo il trattamento.3
- –
Crioterapia. La crioterapia può essere usata in lesioni solitarie che misurano ≤1cm, in particolare nelle regioni acrali, con applicazioni che durano da 30 a 60 secondi. Sia la chirurgia che la crioterapia hanno il vantaggio di poter essere ripetute con buoni risultati.
- –
Terapia intralesionale. Il trattamento intralesionale con vinblastina (0,2mg/mL ogni 2 settimane), vincristina (0,03-0,08mg), o interferone alfa (3-5MIU/3 volte a settimana per 4-5 settimane),10 applicazione topica di alitretinoina 0,1% gel (lesioni maculari) o imiquimod 5% crema (3 volte a settimana per 24 settimane)10 sono trattamenti raccomandati in letteratura, sebbene ci sia una limitata esperienza con questi schemi.5
-
Malattia disseminata
- –
Doxorubicina liposomiale pegilata (PLD) (20mg/m2 ogni 3 settimane). La PLD è il trattamento di scelta tranne che nei pazienti con malattie cardiache. La risposta è buona o eccellente, con risposte parziali (diminuzione del 50% del numero di lesioni tumorali) o risposte complete della durata di 25 mesi in più del 70% dei pazienti.11 La durata della chemioterapia non è ben stabilita, anche se si raccomanda di mantenere il trattamento per 1-2 cicli dopo la risposta clinica. Il trattamento con PLD è generalmente ben tollerato, con effetti collaterali limitati, ed è meno cardiotossico del trattamento con composti tradizionali. Possono quindi essere somministrate dosi cumulative più elevate e trattamenti più lunghi. Le tossicità più gravi (grado 3 e 4) sono poco frequenti e comprendono neutropenia e anemia.12
- –
Vinblastina (3mg/m2/settimana/per via endovenosa o 6mg/m2/2settimana/iv). La vinblastina offre buoni risultati, con tassi di risposta tra il 50% e il 90%, anche se può verificarsi leucopenia.3,13
- –
Altre chemioterapie associate ad alti tassi di risposta, ma con effetti avversi includono paclitaxel iv (100mg/settimana), bleomicina (15U/settimana per 3 settimane e poi ogni 3 settimane/intramuscolo), ed etoposide orale (100mg/die, 3-5 giorni a settimana).10 C’è solo 1 studio clinico randomizzato che ha confrontato la vinblastina iv con l’etoposide orale; non sono state trovate differenze significative nella risposta o nella sopravvivenza, ma è stata riportata una percentuale maggiore di effetti avversi con il trattamento con etoposide.13
Algoritmo per il trattamento del sarcoma di Kaposi.
| Variante | Trattamento | ||
|---|---|---|---|
| Classic KS | Trattamento di scelta | Altre alternative | |
| I. Lesioni solitarie o isolate | a. Considerare solo l’osservazione clinica. b. Radioterapia locale o escissione chirurgica |
Terapia intralesionale | |
| II. Malattia disseminata | Doxorubicina liposomiale | Vinblastina, paclitaxel, etoposide | |
| Sarcoma di Kaposi associato all’HIV | I. Lesioni solitarie o isolate | Iniziare la HAART con o senza terapia locale (radioterapia, escissione chirurgica o crioterapia) | HAART e terapia intralesionale |
| II. Malattia disseminata | HAART e doxorubicina liposomiale | HAART con paclitaxel | |
| KS iatrogeno (associato a immunosoppressione) | Sospendere o ridurre la dose dell’immunosoppressore | Seguire il regime per il trattamento di HIV-KS associato all’HIV | |
Abbreviazioni: HAART, terapia antiretrovirale altamente attiva; KS, sarcoma di Kaposi
Sarcoma di Kaposi associato all’HIV
-
Lesioni solitarie o limitate
- –
HAART in monoterapia o associato a terapia locale (Fig. 4). La HAART viene somministrata come trattamento iniziale, dato che tali agenti hanno dimostrato di ricostituire la funzione immunitaria e di abbassare l’incidenza e la gravità del sarcoma,2,7 con riduzione o scomparsa delle lesioni. Nelle lesioni sintomatiche e antiestetiche, la chirurgia,14 l’elettrocoagulazione o la crioterapia sono opzioni. La vinblastina intralesionale15 (da 0.2 a 0.3mg/mL, somministrando 0.1mL per ogni lesione di 0.5cm2) o la radioterapia a bassa energia (100kV, dosi tra 8Gy/1frazione o 30Gy/10frazioni, più del 95% di risposta clinica completa) possono essere considerate.16 La sopravvivenza libera da lesioni a 5 anni dopo il trattamento HAART, con o senza terapia locale, è stata del 92% in una serie di più di 400 casi.14
 Figura 4.
Figura 4.Algoritmo di trattamento per il sarcoma di Kaposi associato all’HIV.
Abbreviazioni: HAART, terapia antiretrovirale altamente attiva; IRS, sindrome da ricostituzione immunitaria.
Adattato dal Consensus Group for treatment of HIV-associated Kaposi sarcoma. Riunione di consenso. Barcellona: Saned; 1998.
(0.12MB).Malattia Disseminata
Il trattamento sistemico è raccomandato in quei pazienti trattati con HAART e con esteso coinvolgimento cutaneo (più di 15-25 lesioni), gonfiore intenso, KS cutaneo che non ha risposto alla terapia locale o che sta progredendo rapidamente, KS associato alla sindrome da ricostituzione immunitaria, o coinvolgimento sintomatico degli organi interni.
- –
PLD (20mg/m2 ogni 3 settimane). La HAART e la PLD dovrebbero essere iniziate contemporaneamente,17 poiché la combinazione è più efficace della HAART da sola.18 Diversi cicli di trattamento sono solitamente somministrati a seconda della risposta clinica. La risposta completa/parziale si ottiene con la terapia combinata nell’80%,19 con una sopravvivenza a 5 anni superiore all’85%. Le ricadute sono limitate (13%) e si verificano nel primo anno dopo la fine del trattamento.20 La risposta della PLD è superiore alla combinazione di bleomicina, vincristina o vinblastina e doxorubicina non liposomiale,21 e daunorubicina liposomiale.22
- –
Paclitaxel (100mg/m2 ogni 2 settimane). Paclitaxel è un trattamento di seconda linea con risposte favorevoli nel 71% dei pazienti,23 ma con tassi di sopravvivenza inferiori alla PLD e tassi più elevati di tossicità di grado 3-5.14,24 È necessaria la premedicazione con desametasone e possono verificarsi gravi interazioni farmaco-farmaco con gli agenti antiretrovirali.
- –
Altre terapie. Altri farmaci, come l’etoposide, la vinorelbina, l’interleuchina12, il bevacizumab e l’imatinib sono stati usati, ma l’esperienza è limitata.5
Immunosoppressore associato al sarcoma di Kaposi
- –
Sospendere l’immunosoppressore o ridurre la dose. La sospensione o la riduzione della dose di immunosoppressore può indurre remissioni spontanee. Se questo è inefficace o impraticabile, vengono solitamente applicati gli approcci utilizzati nel KS associato all’HIV.5
Conclusioni
KS è un tumore angioproliferativo con diversi sottotipi associati all’età avanzata, a certe popolazioni africane, all’immunosoppressione iatrogena o all’infezione da HIV. Sebbene la HAART abbia portato ad una drastica diminuzione dell’incidenza e della gravità del KS in individui con infezione da HIV, è importante essere consapevoli delle diverse opzioni terapeutiche a seconda della variante di KS e della sua presentazione clinica.
Angiosarcoma cutaneo
Gli angiosarcomi rappresentano tra l’1% e il 2% di tutti i sarcomi, ma almeno la metà sono cutanei.25,26 Tra i sarcomi cutanei, l’angiosarcoma è il quarto più frequente, dopo il KS, il dermatofibrosarcoma e il leiomiosarcoma. L’angiosarcoma cutaneo è una delle neoplasie cutanee con prognosi peggiore, con una sopravvivenza a 5 anni tra il 10% nelle serie più vecchie27 e il 30%-50% in quelle più recenti.25,28,29 Ci sono 3 varianti principali di angiosarcomi cutanei: lesioni idiopatiche sul viso e sul cuoio capelluto di pazienti anziani (angiosarcoma di Wilson-Jones), una variante che rappresenta circa il 50% degli angiosarcomi cutanei, e 2 forme di angiosarcoma secondario, una localizzata in aree di linfedema cronico, in particolare nelle braccia di donne sottoposte a mastectomia radicale (sindrome di Stewart-Treves) e un’altra che si sviluppa su aree di pelle irradiata, in particolare nell’area pettorale di donne sottoposte a radioterapia dopo un cancro al seno (Fig. 5). Si tratta di un sarcoma cutaneo molto aggressivo con frequenti recidive locali e prognosi infausta.27,30 L’unica terapia potenzialmente curativa è la chirurgia con o senza radioterapia.
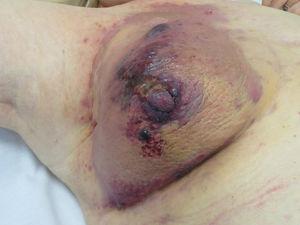 Angiosarcoma. Papule e noduli rosso-violacei su una placca eritemato-violacea su un seno precedentemente irradiato per cancro.Figura 5.
Angiosarcoma. Papule e noduli rosso-violacei su una placca eritemato-violacea su un seno precedentemente irradiato per cancro.Figura 5.Angiosarcoma. Papule e noduli rosso-violacei su una placca eritemato-violacea su un seno precedentemente irradiato per il cancro.
(0.07MB).Epidemiologia e diagnosi clinicaL’incidenza dell’angiosarcoma cutaneo è difficile da calcolare, ma gli angiosarcomi insieme sono riportati ad un tasso di 0,4 casi per milione di abitanti negli Stati Uniti.31 Gli angiosarcomi cutanei rappresentano tra il 35% e il 60% di tutti gli angiosarcomi, con un’incidenza approssimativa di 0,2 casi per milione di abitanti. Predominano nei pazienti anziani con un’età media di 73 anni. Queste lesioni sono molto rare nei bambini o nei pazienti giovani. Il classico angiosarcoma di Wilson-Jones predomina negli uomini e dopo la radioterapia nelle donne.29 Inoltre, colpisce prevalentemente i bianchi.25,29 La maggior parte degli angiosarcomi idiopatici sono localizzati alla testa e al collo (62%), le lesioni post-radiazione predominano sul tronco (24%), in particolare nella zona pettorale (post-radiazione del seno) e dopo il linfedema sugli arti (11%). La maggior parte degli angiosarcomi post-radiazione compare dopo la radioterapia per il cancro della mammella,32 ma ci sono casi in altre aree irradiate e non solo a causa di processi maligni. Il tempo di latenza tra la radioterapia e lo sviluppo dell’angiosarcoma varia, con una media di 5 anni per le sedi mammarie e 10 anni per le altre sedi. Gli angiosarcomi dovuti al linfedema si verificano prevalentemente in aree di linfedema cronico sulle braccia di donne sottoposte a mastectomia radicale (sindrome di Stewart-Treves),33 ma sono stati riportati casi di lesioni sopra il linfedema di qualsiasi eziologia. Il tempo tra la presentazione del linfedema e lo sviluppo dell’angiosarcoma varia notevolmente tra 1 anno e 30 anni.
La forma iniziale più caratteristica è una lesione simile a un livido, a volte edematosa, con una frequenza poco definita, che tende a passare inosservata inizialmente, in particolare sul cuoio capelluto dei pazienti con capelli.34 Nel caso dell’angiosarcoma della testa e del collo, la vera estensione della lesione può essere meglio apprezzata se il paziente tiene la testa sotto il livello del cuore per alcuni secondi. Questa manovra renderà più visibile la parte subclinica che assume un tono violaceo e un aspetto edematoso.35 Più tardi nel corso del decorso clinico, compaiono papule e noduli, occasionalmente con ulcerazione e sanguinamento nelle fasi avanzate. Alcuni casi si presentano direttamente con papule e noduli senza quasi nessuna fase precedente simile a un’ecchimosi. La dimensione media alla diagnosi è di 3-5 cm.28,29 Gli angiosarcomi possono apparire come un tumore solitario o multifocale e la lesione spesso si estende oltre i limiti clinicamente apprezzabili. Il sospetto clinico di angiosarcoma cutaneo deve essere confermato dalla biopsia.
Diagnosi istologica
I 3 tipi di angiosarcoma cutaneo hanno caratteristiche istopatologiche sovrapponibili. Gli angiosarcomi ben differenziati mostrano lumi vascolari rivestiti da cellule endoteliali appiattite, fasci di collagene sezionati, con limitata atipia cellulare. La diagnosi istologica è complessa in queste fasi ed è utile riconoscere alcuni endoteli con cellule prominenti, pleomorfe con nuclei ipercromatici che tendono a sporgere e a creare delle papille, con diversi strati di cellule endoteliali nelle lumine vascolari.36,37 I condotti vascolari sono irregolari e tendono a formare canali anastomizzanti (Fig. 6). Nei casi peggio differenziati, le cellule tumorali sono epitelioidi o fusiformi, con marcata atipia e abbondante mitosi e un modello di crescita più solido con pochi lumi vascolari, tanto che possono essere confuse con il carcinoma o anche con i melanomi o i fibrosarcomi. La presenza di vacuoli intracitoplasmatici come espressione della differenziazione vascolare primitiva può essere molto utile per sospettare una diagnosi corretta in questi casi. L’infiltrazione in caso di angiosarcomi indifferenziati è molto distruttiva e le componenti normali del derma e degli annessi cutanei scompaiono. Un infiltrato infiammatorio a chiazze accompagna spesso il processo e questo infiltrato può a volte essere così denso da assomigliare ad un linfoma.38 L’estensione dei globuli rossi e dell’emosiderina che li accompagnano è molto variabile. Nello stesso angiosarcoma si possono spesso trovare diversi gradi di differenziazione. L’epidermide può essere normale, atrofica o ulcerata. Il grado di differenziazione degli angiosarcomi cutanei non è tradizionalmente pensato per avere un impatto sulla prognosi, e quindi, a differenza di altri sarcomi, il grado istologico non viene preso in considerazione per la stadiazione.39 Questo approccio potrebbe cambiare in futuro, dato che un recente studio con la più grande serie di angiosarcomi cutanei e angiosarcomi dei tessuti molli pubblicata fino ad oggi, con 821 pazienti, ha sviluppato un modello prognostico per la sopravvivenza che includeva il grado istopatologico.25
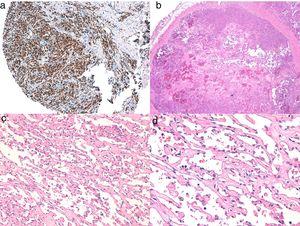 A, Immunoistochimica di angiosarcoma cutaneo positivo per ERG (tipicamente con un pattern nucleare). B, Angiosarcoma con predominanza di aree con pattern vasoformativo. C, D, Immagini dettagliate delle cellule endoteliali neoplastiche, che in questo caso sono prominenti ma senza atipie degne di nota.Figura 6.
A, Immunoistochimica di angiosarcoma cutaneo positivo per ERG (tipicamente con un pattern nucleare). B, Angiosarcoma con predominanza di aree con pattern vasoformativo. C, D, Immagini dettagliate delle cellule endoteliali neoplastiche, che in questo caso sono prominenti ma senza atipie degne di nota.Figura 6.A, Immunoistochimica di un angiosarcoma cutaneo positivo per ERG (tipicamente con un pattern nucleare). B, Angiosarcoma con predominanza di aree con pattern vasoformativo. C, D, Immagini dettagliate delle cellule endoteliali neoplastiche, che in questo caso sono prominenti ma senza atipie degne di nota.
(0.71MB).Lo studio dell’angiosarcoma dovrebbe essere completato con un pannello immunoistochimico che include un pannello di base per i tumori a cellule fusate (CD31, panctocheratine, S110 e actina) e ulteriori marcatori vascolari (CD34, erythroblast transformation-specific-related gene, podoplanina). Alcuni casi di angiosarcoma con predominanza di cellule epitelioidi possono essere positivi per le citocheratine; tuttavia, la positività per i marcatori vascolari come CD31, ERG, e/o podoplanina può escludere un carcinoma indifferenziato. Negli ultimi anni, è stato dimostrato che molti angiosarcomi hanno amplificazione/sovraespressione di MYC. Nella maggior parte degli studi, l’amplificazione di MYC si trova tra il 50% e il 100% degli angiosarcomi secondari ma non generalmente in quelli idiopatici,40-42 ma l’amplificazione o la sovraespressione di MYC è stata rilevata anche in alcuni angiosarcomi idiopatici.43,44 Tuttavia, quasi tutti gli studi hanno dimostrato l’assenza di amplificazione o sovraespressione di MYC nelle proliferazioni vascolari atipiche post-radiazione, e quindi la positività in un caso dubbio di proliferazione vascolare nell’area irradiata quasi esclude l’angiosarcoma.
L’origine dell’angiosarcoma nei vasi sanguigni o linfatici è oggetto di dibattito. L’espressione di CD31 o CD34 è maggiore nelle aree più differenziate, ma in nessuno dei due casi è costante.45 I marcatori immunoistochimici relativamente specifici per i vasi linfatici, come podoplanina, D2-40, LYVE-1, e PROX-1, sono solitamente positivi negli angiosarcomi cutanei, spesso espressi con un pattern immunoistochimico misto di vasi sanguigni endoteliali e vasi linfatici endoteliali.38,46,47 Anche se eccezionali, sono stati riportati casi di angiosarcomi cutanei che esprimono la proteina S-100,48 o marcatori neuroendocrini.49
Staging
Anche se non ci sono linee guida per la gestione dell’angiosarcoma cutaneo, dato che la sede più frequente di metastasi è il polmone50 seguito dai linfonodi,29,51 la tomografia computerizzata toracoaddominale è solitamente raccomandata dopo la diagnosi patologica. Questo studio di imaging deve includere la regione cervicale nel caso di un angiosarcoma della testa e del collo e la pelvi nel caso di angiosarcomi addomino-pelvici post-radiazione. La presenza di disseminazione regionale o a distanza non è rara negli angiosarcomi, ed è riportata rispettivamente nel 30% e nel 10% dei casi.29
Non esiste una stadiazione TNM specifica per gli angiosarcomi, e quindi si utilizza la stadiazione TNM per i sarcomi dei tessuti molli della classificazione dell’American Joint Committee on Cancer, adattata agli angiosarcomi. Nel caso degli angiosarcomi, dato che il grado istologico non è attualmente considerato influente sulla prognosi, gli stadi IA e IIIA sono raggruppati in alcuni studi con gli stadi IB e IIB, che in altri sarcomi sono differenziati solo dal grado istologico.
Trattamento
L’unico trattamento dimostrato essere potenzialmente curativo nell’angiosarcoma cutaneo è la chirurgia, anche se la cura avviene solo sporadicamente. Tuttavia, nei casi inoperabili o metastatici, la radioterapia e/o la chemioterapia hanno un ruolo palliativo riconosciuto. Inoltre, alcuni studi recenti hanno incluso anche la radioterapia come adiuvante alla chirurgia nei casi localizzati di angiosarcoma,50,51 e alcuni autori hanno anche raccomandato di irradiare i linfonodi regionali,52 ma questa non è una pratica comune. Il problema della chirurgia nell’angiosarcoma cutaneo è a volte il carattere multicentrico e la scarsa delimitazione clinica in altri, oltre al fatto che spesso vengono diagnosticati quando le lesioni superano i 5 cm di dimensione. Oltre a questi fattori, i pazienti sono spesso anziani, il che rende più difficile ottenere margini chirurgici adeguati. In generale, se le caratteristiche del tumore e lo stato generale del paziente lo permettono, il trattamento dell’angiosarcoma cutaneo è l’escissione chirurgica con margini sufficienti. Il più accettato è la chirurgia con 3 cm di margine rispetto ai limiti clinicamente apprezzabili.53 La profondità del margine non è ben stabilita, dato che si tratta di un sarcoma dermico, anche se sembrerebbe ragionevole raggiungere le fasce senza asportarle. Nei casi più infiltranti, tuttavia, il muscolo dovrebbe essere incluso per ottenere margini netti. I margini con coinvolgimento dell’angiosarcoma hanno dimostrato di essere un fattore di cattiva prognosi in diversi studi.28,30,51 Nel caso di coinvolgimento del seno, la maggior parte degli studi suggerisce la mastectomia totale o escissioni più o meno estese della pelle irradiata. In casi complessi, la mappatura bioptica preventiva può aiutare la pianificazione prechirurgica. Quando possibile, in oncologia cutanea, la chiusura diretta, gli innesti o le chiusure di seconda intenzione sono preferite per facilitare il follow-up e non mascherare una possibile recidiva locale con la ricostruzione chirurgica, ma questo può essere difficile o impossibile dopo escissioni più radicali di angiosarcomi mammari che richiedono una mastectomia totale con irradiazione dell’intera pelle. Nel caso di angiosarcomi causati da linfedema, uno studio ha rivisto i rapporti di 160 pazienti con sindrome di Stewart-Treves e ha trovato l’assenza di beneficio dell’amputazione rispetto alla chirurgia radicale (con margine di 2 o 3 cm) in questi casi, e quindi l’amputazione degli arti non sembra essere giustificata in tali angiosarcomi.54
Nei casi in cui la chirurgia è impossibile, cioè le lesioni multicentriche o estese, o quelle che interessano aree che complicano la chirurgia, la radioterapia è il trattamento di scelta.55 La dose di radioterapia per l’angiosarcoma cutaneo è di solito 60 Gy distribuiti in 20 sessioni di 3 Gy ciascuna. Quando viene utilizzata come adiuvante alla chirurgia, le dosi sono simili, tranne quando è indicata per gli angiosarcomi post-radiazione, in cui la dose sarà solitamente inferiore (45-50 Gy).
L’unico ruolo della chemioterapia nell’angiosarcoma è come trattamento palliativo, riservato alle lesioni recidivate o metastatiche non suscettibili di chirurgia. Recentemente è stato anche proposto un ruolo neoadiuvante per la chemioterapia prima della chirurgia nei siti periorbitali.56 I chemioterapici più utilizzati nell’angiosarcoma sono il docetaxel,57,58 il paclitaxel,59 e la doxorrubicina liposomiale,60 ma le attuali linee guida NCCN includono anche la vinorelbina, il sorafenib, il sunitinib e il bevacizumab, sebbene i risultati con questi ultimi 3 agenti antiangiogenici siano stati deludenti. La combinazione con i beta-bloccanti in questa fase del trattamento palliativo può essere di qualche beneficio per il paziente.61,62
Follow-up
Non ci sono linee guida standard per il follow-up degli angiosarcomi cutanei. Nel nostro gruppo, abbiamo uno stretto follow-up clinico, con controlli ogni 3-6 mesi per i primi anni, e poi controlli annuali per 10 anni. In queste visite, esaminiamo tutta la pelle e palpiamo i linfonodi territoriali corrispondenti. Almeno una volta all’anno, eseguiamo un’analisi di laboratorio e uno studio di tomografia computerizzata toracoabdominopelvica.
Conflitti di interesse
Gli autori dichiarano di non avere conflitti di interesse.