視神経は、網膜の神経節細胞層を起点とする約120万本の軸索で構成されています。 視神経の軸索はオリゴデンドロサイトによって厚く髄鞘化されており、傷ついても軸索は再生されません。
視神経は、以下の4つの部分に分かれています。
-
眼内部(1mm)(視神経頭部)
-
眼窩内部(25mm)
-
眼窩内部(5-。9mm)
-
頭蓋内部分(10~16mm)
平均的な視神経乳頭の深さは1mmです。 1.5mm、網膜レベルでの高さは1.8mmで、後方に向かって少しずつ広がっています。
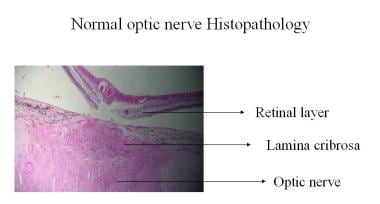 正常な視神経の組織病理学的所見。
正常な視神経の組織病理学的所見。 臨床的には、検眼鏡から入射した光は軸索繊維を介して全内部反射を受け、その後、視板表面の毛細血管で反射することにより、健康な視板に特徴的な黄ピンク色を呈します。
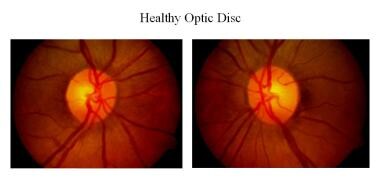 健康な視神経乳頭。
健康な視神経乳頭。 視神経乳頭部の血液供給は、ジン-ハラーの輪から発生する毛細血管によって行われています。 これらの毛細血管は自動調節機能を持ち、漏れはない。 また、これらの毛細血管が失われると、円盤が青白く見える。 Kestenbaum毛細血管数指標は、視神経乳頭部に観察される毛細血管の数である。 正常値は約10本である。 視神経萎縮症ではこの毛細血管の数が6本以下に減少し、12本以上になると充血したディスクになる。
視神経萎縮症で認められる病理学的変化には以下のようなものがあります。
-
ミエリンと軸索の両方の収縮または消失
-
グリオシス
-
生理的カップの深化と篩状層の棒状化
li
-
余分な硬膜を伴うくも膜下腔の拡大
-
孔隔膜の拡大
-
切断された神経は球根状の軸索の膨らみ(Cajal end bulbs)をもたらす。
分類
眼球萎縮症は病的なもの、眼球鏡的なもの、病因的なものに分類されます。
Pathologic optic atrophy
Anterograde degeneration (Wallerian degeneration)
変性は網膜から始まり、側副腎皮質に向かって進行する(例:中毒性網膜症、慢性単純緑内障)。
逆行性変性
変性は軸索の近位部から始まり、視神経に向かって進行する(例:頭蓋内腫瘍による視神経の圧迫)。
トランス・シナプス変性症
トランス・シナプス変性症では、シナプスの一方の側のニューロンが、もう一方の側のニューロンを失った結果、変性します(例:子宮内または乳児期に後頭部に損傷を受けた人)。
眼科的視神経萎縮症
原発性視神経萎縮症
原発性視神経萎縮症の状態(例:下垂体腫瘍、視神経腫瘍、外傷性視神経症、多発性硬化症)では、視神経線維は整然と変性し、視神経頭部の構造に変化はなく、グリア細胞の列に置き換わります。 視神経乳頭の構造は変化せず、視神経線維は整然と変性し、グリア細胞に置き換わります。視神経乳頭は白亜色で境界が鮮明で、網膜血管は正常です。
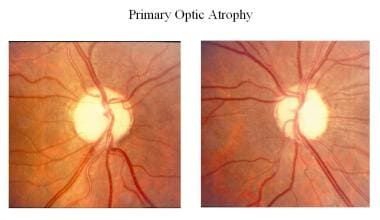 Primary optic atrophy.
Primary optic atrophy. 続発性視神経萎縮症
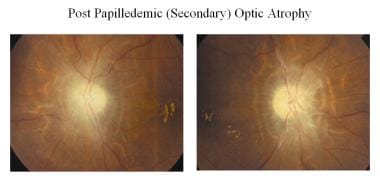
続発性視神経萎縮症(例:乳頭腫、乳頭炎)の状態では、乳頭腫に続発する萎縮が見られます(下図参照)。 視神経線維は著しい変性を示し、グリア組織が過剰に増殖している。 建造物は失われ、その結果、境界が不明瞭になります。 椎間板は灰色または汚れた灰色で、縁は不鮮明であり、増殖した線維膠組織のために篩状層が不明瞭である。 ヒアリン体(corpora amylacea)やドルーゼンが観察されることもあります。 また、毛細血管の鞘や静脈の曲がりくねった部分が見られることもあります。
連続性視神経萎縮症
連続性視神経萎縮症とは、通常、脈絡膜または網膜の疾患に起因する上行型の萎縮症(例:脈絡膜炎、色素性網膜ジストロフィー、黄斑変性症)のことです。
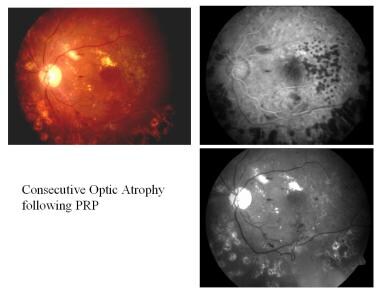 汎網膜光凝固術(PRP)後の連続した視神経萎縮です。
汎網膜光凝固術(PRP)後の連続した視神経萎縮です。 緑内障性視神経萎縮症
空洞性視神経萎縮症としても知られており、緑内障性視神経萎縮症ではディスクの顕著なカップ化が観察されます。 特徴としては、カップの垂直方向の拡大、層状孔の視認(層状ドットサイン)、篩骨膜の後方への反り返り、網膜血管の湾曲と鼻側への移動、乳頭周囲のハローと萎縮などが挙げられる。 椎間板縁のスプリンター出血が観察されることもあります。
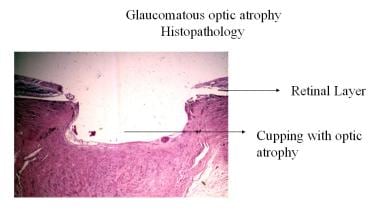
緑内障性視神経萎縮症の病理組織。
側頭部の蒼白
側頭部の蒼白は外傷性視神経症や栄養性視神経症で見られることがあり、多発性硬化症の患者、特に視神経炎の既往のある患者に最もよく見られます。 椎間板は青白く、境界がはっきりしており、血管も正常で、生理的な青白さは時間的にもっとはっきりしています。
病因性視神経萎縮症
遺伝性視神経萎縮症
これは、先天性または乳児性視神経萎縮症(劣性または優性型)、ベーア遺伝性視神経萎縮症(常染色体劣性型)、レーバー視神経萎縮症に分けられます。
常染色体優性の1型視神経萎縮症は、染色体3q29にあるOPA1遺伝子の突然変異によって引き起こされます。 産生されるOPA1タンパク質は、酸化的リン酸化と呼ばれるプロセスや、細胞の自己破壊(アポトーシス)において重要な役割を果たしています。 OPA1は、ミトコンドリアの内膜に存在する不可欠なプロフュージョンタンパク質である。 OPA1遺伝子の変異は、目から脳へ視覚情報を伝達する構造が破壊された人が経験する視覚障害につながる。 OPA1遺伝子の変異により、目から脳へ視覚情報を伝達する構造が破壊されると、まず網膜の神経細胞(網膜神経節細胞)が徐々に失われていきます。
X-linked optic atrophy type 2は、細胞遺伝学的位置Xp11.4-p11.21にあるOPA2遺伝子の突然変異によって引き起こされます。
Hereditary optic atrophy type 3は、細胞遺伝学的位置19q13.32にあるOPA3遺伝子の突然変異が原因です。
3型遺伝性視神経萎縮症は、細胞遺伝学的位置19q13.32にあるOPA3遺伝子の変異によって引き起こされます。 また、III型メチルグルタコン酸尿症との関連もあります。
レーバー遺伝性視神経症は、mtDNA 11778G>A、14484T>C、または3460G>Aのミトコンドリアの点変異から生じます。
連続性萎縮症
連続性萎縮症とは、通常、脈絡膜や網膜の疾患に続いて起こる上行性の萎縮症(例:脈絡膜炎、色素性網膜ジストロフィー、黄斑変性症)のことです。
循環性萎縮(血管性)
循環性とは、毛様体の灌流圧が眼圧を下回るときに見られる虚血性視神経症である。
代謝性萎縮
甲状腺眼症、若年性糖尿病、栄養性弱視、中毒性弱視、タバコ、メチルアルコール、薬物(エタンブトール、スルフォンアミドなど)などの疾患で見られる。
脱髄性萎縮
多発性硬化症やデビック病などの疾患で見られる。
圧迫性または牽引性萎縮
緑内障や乳頭浮腫などの疾患で見られる。
炎症後の萎縮
視神経炎、髄膜の炎症による二次的な脳周囲炎、副鼻腔や眼窩のセルライトなどの疾患で見られるものである。
外傷性視神経症
外傷性視神経症の正確な病態生理はよくわかっていませんが、視神経の剥離や横断、視神経鞘の血腫、異物や骨片の侵入による視神経のインピンジメントなどは、いずれも視神経萎縮につながる外傷性の視神経機能障害を反映しています。
病因にかかわらず、視神経萎縮は様々な程度の視覚障害を伴い、視神経機能検査の1つまたはすべてによって検出されます(「その他の検査」を参照)。
放射線性視神経症
放射線性視神経症は、少なくとも5,000センチグレイの放射線量でより頻繁に起こります。 放射線視神経症は、視神経の血管系や視神経実質の放射線障害に起因する可能性があります。