Kaposi Sarcoma
Kaposi sarcoma (KS) is een angioproliferatieve tumor geassocieerd met het humaan herpesvirus type 8 virus (HHV-8).1,2 Er zijn vier varianten beschreven (tabel 1).
Epidemiologie en klinische kenmerken van de verschillende typen Kaposi-sarcoom.
| Klassieke KS | Endemische KS | HIV-Associated Kaposi Sarcoma | Immunosuppressie-Associated Kaposi Sarcoma | |
|---|---|---|---|---|
| Leeftijd | >60 jaar | 30-45 jaar | 20-50 jaar | 60 jaar |
| Risicogroepen | Mediterrane en Joodse afkomst | Afrikaanse bevolking | HIV-infectie en MSM | Transplantatie-ontvangers, auto-immuunziekten | Plaats | Lenen | Cephalisch, oraal, visceraal | Limben |
| Extracutane manifestaties | Infrequent | Vaak lymfoadenopathisch | Frequent | Mogelijk | Uitkomst | Indolent beloop | Progressief | Angressief, kan worden vertraagd met antiretrovirale therapie | Variabel, kan regressief verlopen met verminderde immunosuppressie |
Afkortingen: MSM, mannen die seks hebben met mannen; KS, Kaposi sarcoom.
Klassiek Kaposi sarcoom. Dit is een zeldzame tumor die meestal voorkomt bij mannen1,3 in het Middellandse-Zeegebied of Midden-Europa, met een incidentie tussen 0,18 en 13,2 gevallen/miljoen.4 Het komt vaker voor bij mannen met chronisch beenoedeem, diabetes mellitus, en corticosteroïdengebruikers. De laesies presenteren zich als enkelvoudige of meervoudige traaggroeiende erythemateuze-violateuze plaques of knobbels, in zeldzame gevallen geassocieerd met lymfoedeem in de extremiteiten en gastro-intestinale en lymfeklierbetrokkenheid. De ziekte heeft een indolent klinisch beloop en 2% van de patiënten overlijdt aan gedissemineerde ziekte.
Endemisch Kaposi-sarcoom. Deze variant is gemeld in equatoriaal Afrika en treft jongvolwassenen en prepuberale personen. Bij volwassenen volgt de variant een indolent of agressief beloop, met betrokkenheid van subcutaan en botweefsel, terwijl de variant zich bij kinderen manifesteert in een agressieve vorm met gegeneraliseerde lymfeklierbetrokkenheid, betrokkenheid van interne organen en afwezigheid van (of beperkte) huidlaesies.5
Immunosuppressie-geassocieerd Kaposi-sarcoom (iatrogene variant). Deze variant is gerapporteerd bij patiënten die met immunosuppressiva worden behandeld en in het bijzonder bij ontvangers van transplantaten. Het risico op het ontwikkelen van KS wordt geschat tussen 150 en 200 keer groter te zijn dan in de algemene bevolking, met een gemiddelde tijd tot begin van 18 maanden.6
HIV-geassocieerd Kaposi sarcoom (epidemische variant). Deze variant is gemeld bij mannen met een hiv-infectie die seks hebben met mannen (MSM-HIV+). Vóór het tijdperk van hoogactieve antiretrovirale therapie (HAART) werd berekend dat 25% van de MSM-HIV+ KS zou ontwikkelen, hoewel dit percentage geleidelijk is gedaald2,7 (Fig. 1 en 2). Bij deze patiënten kan betrokkenheid van huid en mucosa, lymfeklieren, maagdarmkanaal, longen, milt en lever optreden.7 KS bij MSM-HIV- is ook gemeld, in dit geval met een indolent beloop.8
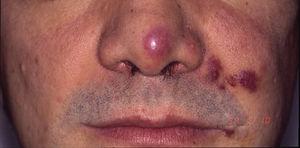
Kaposi-sarcoom bij een patiënt met verworven immunodeficiëntiesyndroom. Erythematisch-violette plaques op het puntje van de neus, in de mondhoek, en op de linkerwang.
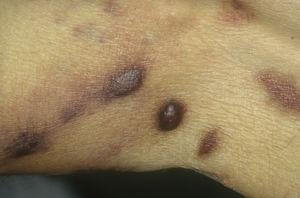
Klassiek Kaposi-sarcoom op de benen. Erythemateuze bruinachtige plaques op het been.
Diagnose
Diagnose wordt klinisch gesteld, maar het is aan te bevelen bevestiging te verkrijgen door biopsie. Histologisch onderzoek toont spindelcellen, proliferatie van onregelmatige vaten met spleetvormige vormen, extravasatie van rode bloedcellen, en leukocytair infiltraat met plasmacellen en intra- en extracellulaire hyalin globules over de dikte van de lederhuid, evenals het zogenaamde promontoriumteken (fig. 3). Polymerasekettingreactie en immunohistochemische kleuring voor latentie-geassocieerd nucleair antigeen (LANA-1) van het HHV-8 virus zijn positief.1
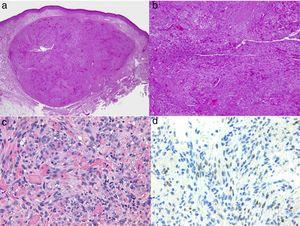
Histologie van Kaposi-sarcoom.
A, Lage-magnificatie beeld van Kaposi-sarcoom in nodulaire fase. Goed gedefinieerde dermale nodule.
B, Sterk cellulaire laesie met enkele openingen in de vorm van scheurtjes.
C, Detail van spindelcellen en rode bloedcellen binnen de kleine vaatjes.
D, Immunohistochemische nucleaire positiviteit voor HHV-8 specifiek voor Kaposi-sarcoom.
Stagering en aanvullende testen
Klassiek Kaposi-sarcoom. Gezien de klinische presentatie (leeftijd, lokale betrokkenheid, infrequente betrokkenheid van inwendige organen, en indolent beloop) is onderzoek van de huid en de lymfeklieren voldoende. Aanvullend onderzoek wordt verricht als de patiënt symptomen vertoont van viscerale aantasting.5
Immunosuppressie-geassocieerd Kaposi sarcoom. Net als voor HIV-geassocieerde KS, die hieronder wordt besproken, is er geen consensus over de stadiëring van immunosuppressie-geassocieerde KS. Gezien het gebrek aan consensus zijn de onderzoeken en aanbevolen toepasselijke criteria meestal dezelfde als voor HIV-geassocieerde KS.
HIV-geassocieerd Kaposi Sarcoom Er is geen geaccepteerd stadiëringssysteem voor deze variant. Röntgenfoto’s van de borstkas worden aanbevolen en in geval van afwijkingen die wijzen op ademhalingsmoeilijkheden, moet bronchoscopie of CT worden verricht. Ook wordt aanbevolen occult fecaal bloed uit te sluiten; als deze test positief is, moet een spijsverteringsendoscopie worden verricht.
In 1989 stelde het oncologiecomité van de AIDS Clinical Trials Group een stadiëring (tabel 2) voor op basis van uitbreiding van de ziekte, CD4T-celaantallen, en systemische aantasting (opportunistische infecties, B-symptomen zoals koorts, gewichtsverlies of aanhoudende diarree, en een Karnofsky-prestatiestatus van minder dan 70 punten). Uit een prospectieve analyse bleek dat deze variabelen verband hielden met de overleving van de patiënt, met als factoren die in verband werden gebracht met een goede prognose: beperkte ziekte, CD4-lymfocytenaantal van meer dan 150 cellen/mm3, en afwezigheid van systemische aantasting.9
Stagering Voorgesteld door de Oncologiecommissie van de AIDS Clinical Trials Groups.
| Goede prognose (0) | Slechte Prognose (1) | |
|---|---|---|
| Tumor | Tumor alleen aangetroffen in de huid of lymfeklieren, of minimale orale betrokkenheid | Tumor die ulceratie veroorzaakt of veroorzaakt Zwaardere orale betrokkenheid KS laesies in het maagdarmkanaal KS laesies op inwendige organen anders dan lymfeklieren |
| Immuniteit | CD4 ≥ 200/μL | CD4 200/μL | Systemische ziekten | Geen opportunistische infecties of spruw Geen B-symptomen Karnofsky performance status ≥ 70 |
Opportunistische infecties of spruw B-symptomen Karnofsky performance status 70 Andere HIV-geassocieerde ziekten (bijvoorbeeld, neurologische ziekte of lymfoom) |
Afkorting; KS, Kaposi-sarcoom.
Langdurige koorts, nachtelijk zweten, meer dan 10% gewichtsverlies, of diarree die langer dan 15 dagen aanhoudt.
Deze voorstellen voor stadiëring werden gedaan voordat HAART beschikbaar was en omvatten geen virale belasting, zodat de toepassing van dit stadiëringssysteem beperkt is gebleven tot klinische trials.
BehandelingKlassiek Kaposi Sarcoom
Er zijn weinig vergelijkende klinische trials van de verschillende behandelingen voor klassiek KS. Meestal worden dezelfde geneesmiddelen en regimes gebruikt als die welke voor epidemische KS worden toegepast (tabel 3).
-
Solitaire laesies.
- –
Klinische observatie. Gezien de leeftijd van de patiënten en de lage mortaliteit, kan follow-up zonder behandeling een optie zijn. In geval van lymfoedeem wordt elastische compressie aanbevolen.10
- –
Lokale radiotherapie. Laag-energetische radiotherapie (100kV: 8Gy in een enkele toepassing of 20-30Gy als gefractioneerde dosis) is effectief voor geïsoleerde laesies.10 In een retrospectieve analyse van 68 patiënten die met radiotherapie werden behandeld, werd een goede respons waargenomen bij 85% van de patiënten, met complete respons bij 58% en verbetering van de symptomen bij 95%.3
- –
chirurgische excisie. In het geval van laesies die ongemak veroorzaken, zoals die in acrale gebieden, wordt chirurgie aanbevolen. In een studie van chirurgisch geëxcideerde laesies bleken patiënten gemiddeld 15 maanden na de behandeling recidiefvrij te zijn.3
- –
Cryotherapie. Cryotherapie kan worden toegepast bij solitaire laesies van ≤ 1 cm, met name in de acrale regio’s, met toepassingen van 30 tot 60 seconden. Zowel chirurgie als cryotherapie hebben het voordeel dat ze kunnen worden herhaald met goede resultaten.
- –
Intralesionale therapie. Intralesionale behandeling met vinblastine (0,2mg/mL elke 2 weken), vincristine (0,03-0,08mg), of interferon alfa (3-5MIU/3 keer per week gedurende 4-5 weken),10 topische toepassing van alitretinoïne 0,1% gel (maculaire laesies) of imiquimod 5% crème (3 keer per week gedurende 24 weken)10 zijn behandelingen die in de literatuur worden aanbevolen, hoewel er beperkte ervaring met deze schema’s is.5
-
Disseminated Disease
- –
Andere chemotherapieën die in verband worden gebracht met hoge responspercentages, maar met bijwerkingen zijn paclitaxel iv (100mg/week), bleomycine (15U/week gedurende 3 weken en vervolgens om de 3 weken/intramusculair), en oraal etoposide (100mg/dag, 3-5 dagen per week).10 Er is slechts 1 gerandomiseerd klinisch onderzoek dat vinblastine iv met oraal etoposide heeft vergeleken; er werden geen significante verschillen gevonden in respons of overleving, maar er werd een hoger percentage bijwerkingen gerapporteerd met etoposidebehandeling.13
–
Pegylated liposomal doxorubicin (PLD) (20mg/m2 om de 3 weken). PLD is de behandeling van keuze, behalve bij patiënten met hartaandoeningen. De respons is goed of uitstekend, met partiële respons (afname van 50% van het aantal tumorlaesies) of complete respons die 25 maanden aanhoudt bij meer dan 70% van de patiënten.11 De duur van de chemotherapie is niet goed vastgesteld, hoewel wordt aanbevolen de behandeling gedurende 1-2 cycli te handhaven na klinische respons. Behandeling met PLD wordt over het algemeen goed verdragen, met beperkte bijwerkingen, en het is minder cardiotoxisch dan behandeling met traditionele verbindingen. Hogere cumulatieve doses en langere behandelingen kunnen daarom worden toegediend. De ernstigste toxiciteiten (graad 3 en 4) komen weinig voor en omvatten neutropenie en anemie.12
–
Vinblastine (3mg/m2/week/intraveneus of 6mg/m2/2wekelijks/iv). Vinblastine biedt goede resultaten, met responspercentages tussen 50% en 90%, hoewel leukopenie kan optreden.3,13
Algoritme voor de behandeling van Kaposi-sarcoom.
| Variant | Behandeling | |||||
|---|---|---|---|---|---|---|
| Klassieke KS | Behandeling naar keuze | Andere Alternatieven | ||||
| a. Overweeg alleen klinische observatie. b. Plaatselijke radiotherapie of chirurgische excisie. |
Intralesionele therapie. | |||||
| Liposomaal doxorubicine | Vinblastine, paclitaxel, etoposide | HIV-geassocieerd Kaposi-sarcoom | I. Solitaire of geïsoleerde laesies | Begin HAART met of zonder lokale therapie (radiotherapie, chirurgische excisie, of cryotherapie) | HAART en intralesionale therapie | |
| HAART en liposomaal doxorubicine | HAART met paclitaxel | |||||
| Schort de dosis van het immuunsuppressivum op of verlaag deze | Volg het schema voor de behandeling van HIV-geassocieerde KS | Volg het schema voor de behandeling van HIV-geassocieerde KS | geassocieerde KS | |||
Afkortingen: HAART, zeer actieve antiretrovirale therapie; KS, Kaposi-sarcoom
HIV-geassocieerd Kaposi-sarcoom
-
Solitaire of beperkte laesies
- –
HAART in monotherapie of geassocieerd met lokale therapie (fig. 4). HAART wordt als initiële behandeling toegediend, aangezien is aangetoond dat dergelijke middelen de immuunfunctie herstellen en de incidentie en de ernst van het sarcoom verminderen,2,7 met vermindering of verdwijning van de laesies. Bij symptomatische en antisthetische laesies zijn chirurgie,14 elektrocoagulatie of cryotherapie opties. Intralesionale vinblastine15 (0,2 tot 0,3mg/mL, toediening van 0,1mL voor elke laesie van 0,5cm2) of laag-energetische radiotherapie (100kV, doses tussen 8Gy/1fractie of 30Gy/10fracties, meer dan 95% volledige klinische respons) kunnen worden overwogen.16 De laesievrije overleving op 5 jaar na HAART-behandeling, met of zonder lokale therapie, was 92% in een serie van meer dan 400 gevallen.14
 Figuur 4.
Figuur 4.Behandelingsalgoritme voor HIV-geassocieerd Kaposi-sarcoom.
Abbreviaties: HAART, highly active antiretroviral therapy; IRS, immuunreconstitutiesyndroom.
Aangepast van de Consensus Group for treatment of HIV-associated Kaposi sarcoma. Consensusvergadering. Barcelona: Saned; 1998.
(0.12MB). -
Disseminated Disease
Systemische behandeling wordt aanbevolen bij patiënten die met HAART worden behandeld en een uitgebreide huidbetrokkenheid hebben (meer dan 15 tot 25 laesies), hevige zwelling, cutane KS die niet heeft gereageerd op lokale therapie of die snel voortschrijdt, KS geassocieerd met immuunreconstitutiesyndroom, of symptomatische betrokkenheid van inwendige organen.
- –
PLD (20mg/m2 om de 3 weken). HAART en PLD moeten tegelijkertijd worden gestart,17 aangezien de combinatie effectiever is dan HAART alleen.18 Afhankelijk van de klinische respons worden gewoonlijk meerdere behandelingskuren toegediend. Complete/partiële respons wordt verkregen met combinatietherapie in 80%,19 met 5-jaars overleving groter dan 85%. Relapses zijn beperkt (13%) en treden op in het eerste jaar na beëindiging van de behandeling.20 De respons van PLD is hoger dan de combinatie van bleomycine, vincristine, of vinblastine en niet-liposomale doxorubicine,21 en liposomale daunorubicine.22
- –
Paclitaxel (100mg/m2 om de 2 weken). Paclitaxel is een tweedelijnsbehandeling met gunstige responsen bij 71% van de patiënten,23 maar met lagere overlevingspercentages dan PLD en hogere percentages graad 3-5 toxiciteiten.14,24 Premedicatie met dexamethason is vereist en ernstige geneesmiddel-geneesmiddelinteracties kunnen optreden met antiretrovirale middelen.
- –
Andere therapieën. Andere geneesmiddelen, zoals etoposide, vinorelbine, interleukine12, bevacizumab en imatinib zijn ook gebruikt, maar de ervaring is beperkt.5
Immunosuppressie-geassocieerd Kaposi Sarcoom
- –
Suspensie van het immunosuppressivum of verlaging van de dosis. Opschorting of verlaging van de dosis immunosuppressivum kan spontane remissies tot gevolg hebben. Als dit niet effectief of onpraktisch is, worden gewoonlijk de benaderingen toegepast die bij HIV-geassocieerde KS worden gebruikt.5
Conclusies
KS is een angioproliferatieve tumor met verschillende subtypes die geassocieerd zijn met gevorderde leeftijd, bepaalde Afrikaanse populaties, iatrogene immunosuppressie of HIV-infectie. Hoewel HAART heeft geleid tot een drastische afname van de incidentie en ernst van KS bij HIV-geïnfecteerden, is het belangrijk om op de hoogte te zijn van de verschillende therapeutische opties, afhankelijk van de KS-variant en de klinische presentatie.
Cutaan angiosarcoom
Angiosarcomen vertegenwoordigen tussen 1% en 2% van alle sarcomen, maar minstens de helft is cutaan.25,26 Van de cutane sarcomen is angiosarcoom het vierde meest frequente, na KS, dermatofibrosarcoom en leiomyosarcoom. Cutaan angiosarcoom is een van de cutane neoplasmata met de slechtste prognose, met een 5-jaars overleving tussen 10% in de oudste reeksen27 en 30%-50% in de meest recente.25,28,29 Er zijn 3 hoofdvarianten van cutane angiosarcomen: idiopathische letsels in het gezicht en op de hoofdhuid van oudere patiënten (Wilson-Jones angiosarcoom), een variant die ongeveer 50% van de cutane angiosarcomen uitmaakt, en 2 vormen van secundair angiosarcoom, een die gelokaliseerd is in gebieden met chronisch lymfoedeem, vooral in de armen van vrouwen die een radicale mastectomie hebben ondergaan (Stewart-Treves syndroom) en een andere die zich ontwikkelt op gebieden van bestraalde huid, vooral in de borststreek van vrouwen die radiotherapie hebben ondergaan na borstkanker (Fig. 5). Dit is een zeer agressief cutaan sarcoom met frequent lokaal recidief en slechte prognose.27,30 De enige potentieel curatieve therapie is chirurgie met of zonder radiotherapie.
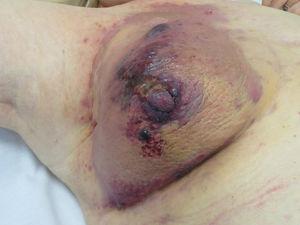
Angiosarcoom. Roodachtige-violette papels en knobbels op een erythemateuze-violette plaque op een borst die eerder bestraald is voor kanker.
Epidemiologie en klinische diagnose
De incidentie van cutane angiosarcomen is moeilijk te berekenen, maar in de Verenigde Staten worden angiosarcomen samen gerapporteerd met een frequentie van 0,4 gevallen per miljoen inwoners.31 Cutane angiosarcomen vertegenwoordigen tussen 35% en 60% van alle angiosarcomen, met een geschatte incidentie van 0,2 gevallen per miljoen inwoners. Ze komen vooral voor bij oudere patiënten met een gemiddelde leeftijd van 73 jaar. Deze laesies zijn zeer zeldzaam bij kinderen of jonge patiënten. Het klassieke Wilson-Jones angiosarcoom overheerst bij mannen en na radiotherapie bij vrouwen.29 Het treft ook overwegend blanken.25,29 Het merendeel van de idiopathische angiosarcomen bevindt zich in het hoofd en de hals (62%), de letsels na de bestraling overheersen op de romp (24%), vooral in de borststreek (na de bestraling van de borst) en na lymfoedeem op de ledematen (11%). De meeste postradiatie angiosarcomen ontstaan na radiotherapie ten gevolge van borstkanker,32 maar er zijn gevallen in andere bestraalde zones en niet alleen ten gevolge van maligne processen. De latentietijd tussen radiotherapie en het ontstaan van angiosarcomen varieert, met een gemiddelde van 5 jaar voor plaatsen in de borst en 10 jaar voor andere plaatsen. Angiosarcomen als gevolg van lymfoedeem komen voornamelijk voor in gebieden met chronisch lymfoedeem op de armen van vrouwen die een radicale mastectomie hebben ondergaan (syndroom van Stewart-Treves),33 maar er zijn gevallen gemeld van laesies boven lymfoedeem van elke etiologie. De tijd tussen presentatie van lymfoedeem en ontwikkeling van angiosarcomen varieert sterk, van 1 jaar tot 30 jaar.
De aanvankelijk meest karakteristieke vorm is een blauwe plek-achtige laesie, soms oedemateus, met een slecht gedefinieerde frequentie, die de neiging heeft aanvankelijk onopgemerkt te blijven, vooral op de hoofdhuid van patiënten met haar.34 In het geval van angiosarcomen van het hoofd en de hals kan de ware omvang van de laesie beter worden beoordeeld als de patiënt zijn of haar hoofd enkele seconden onder het niveau van het hart houdt. Door deze manoeuvre wordt het subklinische deel beter zichtbaar, omdat het een violette toon en een oedemateus uiterlijk aanneemt.35 Later in het klinische beloop verschijnen papels en knobbels, soms met ulceratie en bloedingen in gevorderde fasen. Sommige gevallen presenteren zich direct met papels en knobbels met nauwelijks een voorafgaande blauwe plek-achtige fase. De gemiddelde grootte bij diagnose is 3-5 cm.28,29 Angiosarcomen kunnen verschijnen als een solitaire of multifocale tumor en de laesie zal zich vaak uitbreiden tot buiten de klinisch waarneembare grenzen. Klinische verdenking van cutaan angiosarcoom moet worden bevestigd door biopsie.
Histologische diagnose
De 3 typen cutaan angiosarcoom hebben overlappende histopathologische kenmerken. Goed gedifferentieerde angiosarcomen vertonen vasculaire lumina bekleed door afgeplatte endotheelcellen, dissecterende collageenbundels, met beperkte celatypie. De histologische diagnose is complex in deze fasen en het is nuttig om enkele endotheelcellen te herkennen met prominente, pleomorfe cellen met hyperchromatische kernen die de neiging hebben om uit te steken en enkele papillen te vormen, met meerdere endotheelcellagen in de vasculaire lumina.36,37 De vasculaire conduits zijn onregelmatig en hebben de neiging om anastomoserende kanalen te vormen (Fig. 6). In de slechtst gedifferentieerde gevallen zijn de tumorcellen epithelioïde of fusiform, met duidelijke atypie en overvloedige mitose en een vaster groeipatroon met weinig vasculaire lumina, zodat ze kunnen worden verward met carcinomen of zelfs melanomen of fibrosarcoom. De aanwezigheid van intracytoplasmatische vacuolen als uitdrukking van primitieve vasculaire differentiatie kan in deze gevallen zeer nuttig zijn voor het vermoeden van de juiste diagnose. Infiltratie in het geval van ongedifferentieerde angiosarcomen is zeer destructief en normale componenten van de dermis en huidaanhangsels verdwijnen. Het proces gaat vaak gepaard met een vlekkerig ontstekingsinfiltraat, dat soms zo dicht kan zijn dat het op een lymfoom lijkt.38 De mate van begeleidende rode bloedcellen en hemosiderine is zeer variabel. Verschillende differentiatiegraden kunnen vaak in hetzelfde angiosarcoom worden aangetroffen. De epidermis kan normaal, atrofisch of ulcererend zijn. De differentiatiegraad van cutane angiosarcomen wordt traditioneel niet geacht een invloed te hebben op de prognose, en bijgevolg wordt, in tegenstelling tot bij andere sarcomen, geen rekening gehouden met de histologische graad voor de stadiëring.39 Deze benadering kan in de toekomst veranderen, aangezien in een recente studie met de grootste tot op heden gepubliceerde reeks cutane angiosarcomen en angiosarcomen van weke delen, met 821 patiënten, een prognostisch model voor overleving is ontwikkeld waarin de histopathologische graad is opgenomen.25
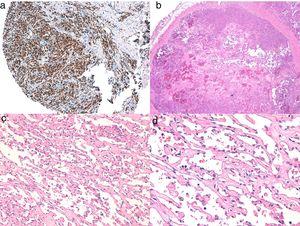
A, Immunohistochemie van cutaan angiosarcoom dat positief is voor ERG (typisch met een kernpatroon). B, Angiosarcoom met predominantie van gebieden met vasoformatief patroon. C, D, Gedetailleerde beelden van neoplastische endotheelcellen, die in dit geval prominent aanwezig zijn, maar zonder noemenswaardige atypie.
Studie van angiosarcomen moet worden aangevuld met een immunohistochemisch panel dat een basispanel voor spindle-celtumoren (CD31, pancytokeratines, S110 en actine) en aanvullende vasculaire markers (CD34, erythroblast transformation-specific gene , podoplanine) bevat. Sommige gevallen van angiosarcomen met overwegend epithelioïde cellen kunnen positief zijn voor cytokeratines; positiviteit voor vasculaire markers zoals CD31, ERG, en/of podoplanine kan echter ongedifferentieerd carcinoom uitsluiten. De laatste jaren is aangetoond dat veel angiosarcomen MYC-amplificatie/overexpressie hebben. In de meeste studies wordt MYC-amplificatie gevonden in tussen 50% en 100% van de secundaire angiosarcomen, maar over het algemeen niet in idiopathische,40-42 maar MYC-amplificatie of -overexpressie is ook gedetecteerd in sommige idiopathische angiosarcomen.43,44 Bijna alle studies hebben echter de afwezigheid van MYC-amplificatie of -overexpressie aangetoond in atypische post-bestraalde vasculaire proliferaties, en dus sluit positiviteit in een twijfelgeval van vasculaire proliferatie in het bestraalde gebied angiosarcomen bijna uit.
De oorsprong van angiosarcomen in bloedvaten of lymfevaten is onderwerp van discussie. Expressie van CD31 of CD34 is groter in de meest gedifferentieerde gebieden, maar is in geen van beide gevallen constant.45 Immunohistochemische markers die relatief specifiek zijn voor lymfevaten, zoals podoplanine, D2-40, LYVE-1, en PROX-1, zijn meestal positief in cutane angiosarcomen, vaak uitgedrukt met een gemengd immunohistochemisch patroon van endotheliale bloedvaten en endotheliale lymfevaten.38,46,47 Hoewel uitzonderlijk, zijn er gevallen bekend van cutane angiosarcomen die S-100 proteïne,48 of neuro-endocriene markers tot expressie brengen.49
Stagering
Hoewel er geen richtlijnen zijn voor de behandeling van cutane angiosarcomen, wordt, gezien het feit dat de meest frequente plaats van metastase de long50 is, gevolgd door de lymfeklieren,29,51 thoracoabdominale computertomografie gewoonlijk aanbevolen na pathologische diagnose. Dit beeldvormingsonderzoek moet de cervicale regio omvatten in het geval van een angiosarcoom van het hoofd en de hals en het bekken in het geval van angiosarcomen van het abdominopelviene type na bestraling. De aanwezigheid van regionale of distante disseminatie is niet ongewoon bij angiosarcomen, en wordt gemeld in respectievelijk 30% en 10% van de gevallen.29
Er is geen TNM-stadiëring specifiek voor angiosarcomen, en daarom wordt de TNM-stadiëring voor weke delen sarcomen van de American Joint Committee on Cancer classificatie, aangepast aan angiosarcomen, gebruikt. Aangezien de histologische graad in het geval van angiosarcomen momenteel niet van invloed wordt geacht op de prognose, worden de stadia IA en IIA in sommige studies gegroepeerd met de stadia IB en IIB, die in andere sarcomen alleen door de histologische graad worden onderscheiden.
Behandeling
De enige behandeling waarvan is aangetoond dat zij bij cutane angiosarcomen mogelijk curatief is, is chirurgie, hoewel genezing slechts sporadisch voorkomt. Inoperabele of gemetastaseerde gevallen hebben radiotherapie en/of chemotherapie echter een erkende palliatieve rol. Bovendien hebben enkele recente studies radiotherapie opgenomen als adjuvans voor chirurgie in gelokaliseerde gevallen van angiosarcoom,50,51 en sommige auteurs hebben zelfs aanbevolen om regionale lymfeklieren te bestralen,52 maar dit is geen gangbare praktijk. Het probleem met chirurgie bij cutane angiosarcomen is soms het multicenter karakter en de slechte klinische afbakening in andere gevallen, evenals het feit dat ze vaak worden gediagnosticeerd wanneer de laesies groter dan 5 cm zijn. Naast deze factoren zijn de patiënten vaak bejaard, wat het moeilijker maakt om geschikte chirurgische marges te verkrijgen. In het algemeen, als de kenmerken van de tumor en de algemene toestand van de patiënt het toelaten, bestaat de behandeling van cutaan angiosarcoom uit chirurgische excisie met voldoende marges. Het meest aanvaard is chirurgie met een marge van 3 cm ten opzichte van de klinisch merkbare grenzen.53 De diepte van de marge is niet goed vastgesteld, gezien het feit dat het om een dermaal sarcoom gaat, hoewel het redelijk lijkt om de fascia te bereiken zonder deze te excideren. In de meest infiltratieve gevallen moet de spier echter worden ingesloten om duidelijke marges te bereiken. In verschillende studies is aangetoond dat marges waarbij angiosarcoom betrokken is, een factor voor slechte prognose zijn.28,30,51 In het geval van borstbetrokkenheid suggereren de meeste studies totale mastectomie of meer of minder uitgebreide excisies van bestraalde huid. In complexe gevallen kan het vooraf in kaart brengen van de biopsie helpen bij de pre-operatieve planning. Waar mogelijk wordt in de cutane oncologie de voorkeur gegeven aan directe sluiting, transplantaten of tweede-intentsluitingen om de follow-up te vergemakkelijken en een mogelijk lokaal recidief met chirurgische reconstructie niet te maskeren, maar dit kan moeilijk of onmogelijk zijn na meer radicale excisies van mamma-angiosarcomen die een totale mastectomie met bestraling van de gehele huid vereisen. In het geval van angiosarcomen ten gevolge van lymfoedeem werden in een studie 160 patiënten met het Stewart-Treves-syndroom onderzocht en werd geen voordeel van amputatie gevonden in vergelijking met radicale chirurgie (met een marge van 2 of 3 cm) in deze gevallen, zodat amputatie van ledematen niet gerechtvaardigd lijkt bij dergelijke angiosarcomen.54
In gevallen waarin chirurgie onmogelijk is, dat wil zeggen multicentrische of uitgebreide laesies, of laesies die gebieden aantasten die chirurgie bemoeilijken, is radiotherapie de behandeling van keuze.55 De dosis radiotherapie voor cutane angiosarcomen is meestal 60 Gy verdeeld over 20 sessies van elk 3 Gy. Wanneer radiotherapie wordt gebruikt als adjuvans voor chirurgie, zijn de doses vergelijkbaar, behalve wanneer de behandeling is geïndiceerd voor angiosarcomen na bestraling, waarbij de dosis gewoonlijk lager zal zijn (45-50 Gy).
De enige rol van chemotherapie bij angiosarcomen is als palliatieve behandeling, gereserveerd voor recidief of metastatische laesies die niet chirurgisch kunnen worden behandeld. Recentelijk is ook een neoadjuvante rol voorgesteld voor chemotherapie voorafgaand aan chirurgie op periorbitale plaatsen.56 De meest gebruikte chemotherapieën bij angiosarcomen zijn docetaxel,57,58 paclitaxel,59 en liposomale doxorrubicine,60 maar de huidige NCCN-richtlijnen omvatten ook vinorelbine, sorafenib, sunitinib en bevacizumab, hoewel de resultaten met deze laatste drie antiangiogene middelen teleurstellend zijn geweest. Combinatie met bètablokkers in deze fase van palliatieve behandeling kan van enig nut zijn voor de patiënt.61,62
Follow-up
Er zijn geen standaardrichtlijnen voor de follow-up van cutane angiosarcomen. In onze groep hebben we een nauwgezette klinische follow-up, met controlebezoeken om de 3-6 maanden gedurende de eerste paar jaar, en vervolgens jaarlijkse controlebezoeken gedurende 10 jaar. Tijdens deze bezoeken onderzoeken we de gehele huid en palperen we de bijbehorende territoriale lymfeklieren. Ten minste eenmaal per jaar voeren we een laboratoriumanalyse en een thoracoabdominopelvisch computertomografie-onderzoek uit.
Belangenconflicten
De auteurs verklaren dat zij geen belangenconflicten hebben.