Kaposi Sarcoma
Kaposi sarcoma (KS) é um tumor angioproliferativo associado ao vírus do herpesvírus humano tipo 8 (HHV-8).1,2 Foram descritas quatro variantes (Tabela 1).
| Classic KS | Endemia KS | HIV-Sarcoma de Kaposi associado | Immunosupressão-Sarcoma Kaposi associado | |
|---|---|---|---|---|
| Age | 30-45 anos | 60 anos | ||
| Grupos de risco | Descendência mediterrânica e judaica | População africana | Infecção pelo HIV e HSH | Receptores de plantas, doenças auto-imunes |
| Site | Pernas | Pernas | Cefálico, oral, visceral | Limbs |
| Infrequente | Limites | Frequente | Possivel | |
| Outcome | Indolent Course | Progressivo | Aggressivo, pode ser atrasado com terapia anti-retroviral | Variável, pode regredir com diminuição da imunossupressão |
A abreviaturas: MSM, homens que fazem sexo com homens; KS, sarcoma de Kaposi.
/div>
Classic Kaposi sarcoma. Trata-se de um tumor invulgar que tende a afectar homens1,3 na região mediterrânica ou da Europa Central, com uma incidência entre 0,18 e 13,2 casos/milhão.4 Ocorre com maior frequência em homens com edema crónico das pernas, diabetes mellitus, e utilizadores de corticosteróides. As lesões apresentam-se como placas ou nódulos eritemato-violáceos de crescimento lento, únicos ou múltiplos, em raras ocasiões associados a linfedema nas extremidades e envolvimento gastrintestinal e linfonodal. Tem um curso clínico indolente, e 2% dos doentes morrem de doença disseminada.
Sarcoma endémico de Kaposi. Esta variante foi notificada na África equatorial e afecta adultos jovens e indivíduos pré-púberes. Em adultos, a variante segue um curso indolente ou agressivo, com envolvimento de tecido subcutâneo e ósseo, enquanto que em crianças se manifesta de forma agressiva com envolvimento generalizado de gânglios linfáticos, envolvimento de órgãos internos, e ausência de (ou limitação de) lesões cutâneas.5
Sarcoma de Kaposi associado à imunossupressão (variante iatrogénica). Esta variante foi relatada em doentes em tratamento com imunossupressores e em receptores de transplante em particular. O risco de desenvolvimento de KS é estimado entre 150 e 200 vezes maior do que na população geral, com um tempo médio de início de 18 meses.6
Sarcoma de Kaposi associado ao VIH (variante epidémica). Esta variante tem sido relatada em homens com infecção por HIV que fazem sexo com homens (HSH-HIV+). Antes da era da terapia anti-retroviral altamente activa (HAART), calculou-se que 25% do MSM-HIV+ desenvolveria KS, embora esta percentagem tenha diminuído progressivamente2,7 (Figs. 1 e 2). Estes doentes podem experimentar envolvimento de pele e mucosa, gânglios linfáticos, tracto gastrointestinal, pulmões, baço e fígado.7 KS em MSM-HIV- também foi relatado, neste caso com um curso indolente.8
 br>>>/div>
br>>>/div>
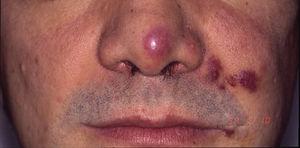
Kaposi sarcoma num doente com síndrome de imunodeficiência adquirida. Placas eritmato-violáceas na ponta do nariz, canto da boca, e bochecha esquerda.
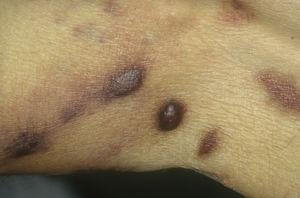
Classic Kaposi sarcoma nas pernas. Placas eritematosas acastanhadas na perna.
Diagnóstico
O diagnóstico é realizado clinicamente, mas recomenda-se a obtenção de confirmação por biopsia. O estudo histológico revela células fusiformes, proliferação de vasos irregulares com formas de fenda, extravasamento de glóbulos vermelhos, e infiltrado leucocitário com plasmócitos e glóbulos hialina intra e extracelulares em toda a espessura da derme, bem como o chamado sinal promontório (Fig. 3). A reacção em cadeia da polimerase e a coloração imunohistoquímica para o antigénio nuclear associado à latência (LANA-1) do vírus HHV-8 são positivas.1
 br>>>div>
br>>>div>
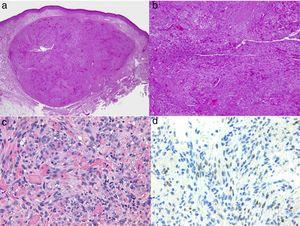
Histologia do sarcoma de Kaposi.
A, visão de baixa magnificação do sarcoma de Kaposi em fase nodular. Nódulo dérmico bem definido.
B, Lesão altamente celular com alguns espaços em forma de fendas.
C, Detalhe das células do fuso e glóbulos vermelhos dentro dos pequenos vasos.
D, Positividade nuclear imunohistoquímica para HHV-8 específica do sarcoma de Kaposi.
Testes de Estágio e Complementares
Classic Kaposi sarcoma. Tendo em conta a apresentação clínica (idade, envolvimento local, envolvimento pouco frequente de órgãos internos, e curso indolente), o exame da pele e dos gânglios linfáticos é suficiente. São realizados testes complementares se o paciente apresentar sintomas de comprometimento visceral.5
Sarcoma de Kaposi associado à imunossupressão. Quanto ao KS associado ao VIH discutido abaixo, não existe um estadiamento consensual para o KS associado à imunossupressão. Dada a falta de consenso, os testes e critérios aplicáveis recomendados são geralmente os mesmos que para o KS associado ao VIH.
HIV-Associated Kaposi Sarcoma Não existe um sistema de estadiamento aceite para esta variante. Recomenda-se a radiografia do tórax e, em caso de anomalias sugestivas de comprometimento respiratório, deve ser realizada a broncoscopia ou tomografia computorizada do tórax. Também é recomendado excluir sangue oculto fecal; se este teste for positivo, deve ser realizada endoscopia digestiva.
Em 1989, o Comité de Oncologia do Grupo de Ensaios Clínicos SIDA propôs o estadiamento (Quadro 2) com base na extensão da doença, contagem de células CD4T, e comprometimento sistémico (infecções oportunistas, sintomas B tais como febre, perda de peso, ou diarreia persistente, e estado de desempenho de Karnofsky abaixo dos 70 pontos). Uma análise prospectiva mostrou que estas variáveis estavam relacionadas com a sobrevivência do paciente, sendo os factores associados a um bom prognóstico a doença limitada, contagem de linfócitos CD4 superior a 150 células/mm3, e ausência de compromisso sistémico.9
| Good Prognosis (0) | Poor Prognosis (1) | |
|---|---|---|
| Tumor | Tumor apenas encontrado na pele ou nos gânglios linfáticos, ou envolvimento oral mínimo | |
| Imunidade | CD4 ≥ 200/μL | CD4 200/μL |
A abreviatura; KS, Kaposi sarcoma.
Febre prolongada, suor nocturno, perda de peso superior a 10%, ou diarreia com duração superior a 15 dias.
/div>
Estas propostas de estadiamento foram apresentadas antes da HAART estar disponível e não incluem a carga viral, pelo que a aplicação deste sistema de estadiamento foi limitada aos ensaios clínicos.
TreatmentClassic Kaposi Sarcoma
Existem poucos ensaios clínicos comparativos dos diferentes tratamentos para a KS clássica. São geralmente utilizados os mesmos medicamentos e regimes que os aplicados para a epidemia de KS (Tabela 3).
- p>p> Lesões solitárias.
- -p> Observação clínica. Dada a idade dos pacientes e a baixa mortalidade, o acompanhamento sem tratamento pode ser uma opção. No caso de linfedema, recomenda-se a compressão elástica.10
- –
Radioterapia local. A radioterapia de baixa energia (100kV: 8Gy numa única aplicação ou 20-30Gy como dose fraccionada) é eficaz para lesões isoladas.10 Numa análise retrospectiva de 68 pacientes tratados com radioterapia, observou-se uma boa resposta em 85% dos pacientes, com resposta completa em 58% e melhoria dos sintomas em 95%.3
- –
Excisão cirúrgica. No caso de lesões que causam desconforto, tais como as que ocorrem em áreas acrílicas, a cirurgia é recomendada. Num estudo das lesões cirurgicamente excisadas, verificou-se que os pacientes ficaram sem recidivas durante uma média de 15 meses após o tratamento.3
- –
Crioterapia. A crioterapia pode ser utilizada em lesões solitárias medindo ≤1cm, particularmente em regiões acrílicas, com aplicações que duram de 30 a 60 segundos. Tanto a cirurgia como a crioterapia têm a vantagem de poderem ser repetidas com bons resultados.
- –
Terapia intra-esional. Tratamento intralesional com vinblastina (0,2mg/mL cada 2 semanas), vincristina (0,03-0,08mg), ou interferon alfa (3-5MIU/3 vezes por semana durante 4-5 semanas),10 aplicação tópica de alitretinoína 0,1% gel (lesões maculares) ou imiquimod 5% creme (3 vezes por semana durante 24 semanas)10 são tratamentos recomendados na literatura, embora haja uma experiência limitada com estes regimes.5
- p>Doença disseminada
- – -p>Doxorubicina lipossomal pegilada (PLD) (20mg/m2 a cada 3 semanas). A PLD é o tratamento de escolha, excepto em doentes com doenças cardíacas. A resposta é boa ou excelente, com respostas parciais (redução de 50% no número de lesões tumorais) ou resposta completa com duração de 25 meses em mais de 70% dos pacientes.11 A duração da quimioterapia não está bem estabelecida, embora seja recomendado manter o tratamento durante 1-2 ciclos após a resposta clínica. O tratamento com PLD é geralmente bem tolerado, com efeitos secundários limitados, e é menos cardiotóxico do que o tratamento com compostos tradicionais. Por conseguinte, podem ser administradas doses cumulativas mais elevadas e tratamentos mais longos. As toxidades mais severas (grau 3 e 4) são pouco frequentes e incluem neutropenia e anemia.12
- –
Vinblastina (3mg/m2/semana/intravenosa ou 6mg/m2/2semana/iv). A vinblastina oferece bons resultados, com taxas de resposta entre 50% e 90%, embora a leucopenia possa ocorrer.3,13
- –
Outras quimioterapias associadas a altas taxas de resposta, mas com efeitos adversos incluem paclitaxel iv (100mg/semana), bleomicina (15U/semana durante 3 semanas e depois a cada 3 semanas/intramuscularmente), e etoposídeo oral (100mg/dia, 3-5 dias por semana).10 Há apenas 1 ensaio clínico aleatório que comparou a vinblastina iv com etoposida oral; não foram encontradas diferenças significativas em resposta ou sobrevivência, mas uma maior proporção de efeitos adversos foi relatada com o tratamento com etoposida.13
| Variante | Tratamento | ||
|---|---|---|---|
| Classic KS | Tratamento de Escolha | Outras Alternativas | |
| I. Lesões solitárias ou isoladas | a. Considerar apenas observação clínica. b. Radioterapia local ou excisão cirúrgica |
Terapia intra-esional | |
| II. Doença disseminada | Doxorubicina lipossomal | Vinblastina, paclitaxel, etoposídeo | |
| HIV-Associated Kaposi Sarcoma | I. Lesões solitárias ou isoladas | Iniciar HAART com ou sem terapia local (radioterapia, excisão cirúrgica, ou crioterapia) | HAART e terapia intralesional |
| II. Doença disseminada | HAART e doxorubicina lipossomal | HAART com paclitaxel | |
| Iatrogénica KS (associada à imunossupressão) | Suspender ou reduzir a dose do imunossupressor | Sigir o regime de tratamento do HIV-associado KS | |
Abreviaturas: HAART, terapia anti-retroviral altamente activa; KS, sarcoma de Kaposi
HIV-Associated Kaposi Sarcoma
- p>> Lesões Solitárias ou Limitadas
- -p>HAART em monoterapia ou associada a terapia local (Fig. 4). HAART é administrado como tratamento inicial, dado que se demonstrou que tais agentes reconstituem a função imunitária e reduzem a incidência e gravidade do sarcoma,2,7 com redução ou desaparecimento das lesões. Nas lesões sintomáticas e antiestéticas, a cirurgia,14 a electrocoagulação, ou a crioterapia são opções. Pode ser considerada a vinblastino intralesional15 (0,2 a 0,3mg/mL, administrando 0,1mL para cada lesão de 0,5cm2) ou radioterapia de baixa energia (100kV, doses entre 8Gy/1fracção ou 30Gy/10fracções, mais de 95% de resposta clínica completa).16 Sobrevida sem lesão aos 5 anos após o tratamento HAART, com ou sem terapia local foi de 92% numa série de mais de 400 casos.14
 br>>>div>
div>div>div>Figure 4.
br>>>div>
div>div>div>Figure 4.algoritmo de tratamento do sarcoma de Kaposi associado ao HIV.
A abreviaturas: HAART, terapia anti-retroviral altamente activa; IRS, síndrome de reconstituição imunitária.
Adaptado do Grupo de Consenso para o tratamento do sarcoma de Kaposi associado ao VIH. Reunião de Consenso. Barcelona: Saned; 1998.
(0.12MB).
p>Disseminated Disease
tratamento sistémico é recomendado nos pacientes tratados com HAART e com extenso envolvimento cutâneo (mais de 15 a 25 lesões), inchaço intenso, KS cutânea que não respondeu à terapia local ou que está a progredir rapidamente, KS associada à síndrome de reconstituição imunitária, ou envolvimento sintomático de órgãos internos.
- –
PLD (20mg/m2 a cada 3 semanas). HAART e PLD devem ser iniciados ao mesmo tempo,17 uma vez que a combinação é mais eficaz do que HAART sozinho.18 Vários cursos de tratamento são geralmente administrados dependendo da resposta clínica. A resposta completa/parcial é obtida com terapia combinada em 80%,19 com 5 anos de sobrevivência superior a 85%. As recidivas são limitadas (13%) e ocorrem no primeiro ano após a conclusão do tratamento.20 A resposta PLD é superior à combinação de bleomicina, vincristina, ou vinblastina e doxorubicina nãolipossomal,21 e daunorubicina lipossomal.22
- –
Paclitaxel (100mg/m2 a cada 2 semanas). O paclitaxel é um tratamento de segunda linha com respostas favoráveis em 71% dos pacientes,23 mas com taxas de sobrevivência inferiores à PLD e taxas mais elevadas de toxicidade de grau 3-5.14,24 É necessária uma pré-medicação com dexametasona e podem ocorrer interacções medicamentosas graves com agentes anti-retrovirais.
- –
Outras terapias. Outros medicamentos, tais como etoposide, vinorelbina, interleukin12, bevacizumab, e imatinib, também têm sido utilizados, mas a experiência é limitada.5
Immunosupressão-Associada ao Kaposi Sarcoma
- –
Suspender o imunossupressor ou reduzir a dose. Suspensão ou redução da dose do imunossupressor pode induzir remissões espontâneas. Se isto for ineficaz ou impraticável, as abordagens usadas no KS associado ao VIH são geralmente aplicadas.5
Conclusões
KS é um tumor angioproliferativo com diferentes subtipos associados à idade avançada, certas populações africanas, imunossupressão iatrogénica, ou infecção pelo VIH. Embora o HAART tenha levado a uma diminuição dramática na incidência e gravidade do KS em indivíduos com infecção pelo HIV, é importante estar ciente das diferentes opções terapêuticas de acordo com a variante KS e a sua apresentação clínica.
Angiossarcoma cutâneo
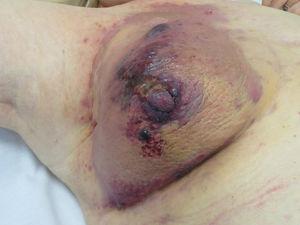
Angiossarcoma representam entre 1% e 2% de todos os sarcomas, mas pelo menos metade são cutâneos.25,26 Dos sarcomas cutâneos, o angiossarcoma é o quarto mais frequente, atrás do KS, do dermatofibrossarcoma, e do leiomiossarcoma. O angiossarcoma cutâneo é uma das neoplasias cutâneas com pior prognóstico, com 5 anos de sobrevivência entre 10% nas séries mais antigas27 e 30%-50% nas mais recentes.25,28,29 Existem 3 variantes principais de angiossarcomas cutâneos: lesões idiopáticas na face e couro cabeludo de pacientes idosos (angiossarcoma de Wilson-Jones), uma variante que representa aproximadamente 50% dos angiossarcomas cutâneos, e 2 formas de angiossarcoma secundário, uma localizada em áreas de linfedema crónico, particularmente nos braços de mulheres que se submetem a mastectomia radical (síndrome de Stewart-Treves) e outra que se desenvolve sobre áreas de pele irradiada, particularmente na área peitoral de mulheres que se submetem a radioterapia após cancro da mama (Fig. 5). Trata-se de um sarcoma cutâneo muito agressivo com frequentes recidivas locais e mau prognóstico.27,30 A única terapia potencialmente curativa é a cirurgia com ou sem radioterapia.
 br>
br>
Angiossarcoma. Pápulas e nódulos vermelhos-violáceos numa placa eritemato-violácea numa mama previamente irradiada para o cancro.
Epidemiologia e Diagnóstico Clínico
A incidência de angiossarcoma cutâneo é difícil de calcular, mas os angiossarcomas em conjunto são notificados a uma taxa de 0,4 casos por milhão de habitantes nos Estados Unidos.31 Os angiossarcomas cutâneos representam entre 35% e 60% de todos os angiossarcomas, com uma incidência aproximada de 0,2 casos por milhão de habitantes. Predominam em doentes idosos com uma idade média de 73 anos. Estas lesões são muito raras em crianças ou doentes jovens. O angiossarcoma clássico de Wilson-Jones predomina nos homens e após radioterapia nas mulheres.29 Também afecta predominantemente os brancos.25,29 A maioria dos angiossarcomas idiopáticos localiza-se na cabeça e pescoço (62%), as lesões pós-tradiação predominam no tronco (24%), particularmente na zona peitoral (pós-tradiação do peito) e após linfedema nos membros (11%). A maioria dos angiossarcomas pós-tradiação aparecem após a radioterapia devido ao cancro da mama,32 mas há casos noutras áreas irradiadas e não apenas devido a processos malignos. O tempo de latência entre a radioterapia e o desenvolvimento do angiossarcoma varia, com uma média de 5 anos para locais de mama e 10 anos para outros locais. Angiossarcoma devido a linfedema ocorre predominantemente em áreas de linfedema crónico nos braços de mulheres que são submetidas a mastectomia radical (síndrome de Stewart-Treves),33 mas têm sido relatados casos de lesões sobre linfedema de qualquer etiologia. O tempo entre a apresentação do linfedema e o desenvolvimento de angiossarcomas varia muito entre 1 ano e 30 anos.
A forma mais característica inicial é uma lesão semelhante a uma contusão, por vezes edematosa, com uma frequência mal definida, que tende a passar despercebida inicialmente, particularmente no couro cabeludo dos pacientes com cabelo.34 No caso de angiossarcoma da cabeça e pescoço, a verdadeira extensão da lesão pode ser melhor apreciada se o paciente segurar a sua cabeça abaixo do nível do coração durante alguns segundos. Esta manobra tornará a parte subclínica mais visível à medida que adquire um tom violento e aspecto edematoso.35 Mais tarde, no curso clínico, aparecem pápulas e nódulos, ocasionalmente com ulceração e hemorragia em fases avançadas. Alguns casos apresentam-se directamente com pápulas e nódulos com praticamente nenhuma fase anterior de hematoma. O tamanho médio no diagnóstico é de 3-5 cm.28,29 Angiossarcomas pode aparecer como um tumor solitário ou multifocal e a lesão irá frequentemente estender-se para além dos limites clinicamente apreciáveis. A suspeita clínica de angiossarcoma cutâneo deve ser confirmada por biopsia.
Diagnóstico histológico
Os 3 tipos de angiossarcoma cutâneo têm características histopatológicas sobrepostas. Os angiossarcoma bem diferenciados mostram lumina vascular revestida por células endoteliais achatadas, dissecando feixes de colagénio, com atipias celulares limitadas. O diagnóstico histológico é complexo nestas fases e é útil reconhecer alguns endotélios com células pleomórficas proeminentes, com núcleos hipercromáticos que tendem a sobressair e a criar algumas papilas, com várias camadas celulares endoteliais na lumina vascular.36,37 Os condutos vasculares são irregulares e tendem a formar canais de anastomose (Fig. 6). Nos casos mais diferenciados, as células tumorais são epitélioides ou fusiformes, com marcada atipia e mitose abundante e um padrão de crescimento mais sólido com pouca lumina vascular, de tal forma que podem ser confundidas com carcinoma ou mesmo melanomas ou fibrossarcomas. A presença de vacúolos intracitoplasmáticos como expressão de diferenciação vascular primitiva pode ser muito útil para a suspeita de um diagnóstico correcto nestes casos. A infiltração no caso de angiossarcomas indiferenciados é muito destrutiva e os componentes normais da derme e dos apêndices cutâneos desaparecem. Um infiltrado inflamatório desigual acompanha frequentemente o processo e este infiltrado pode por vezes ser tão denso que se assemelha a um linfoma.38 A extensão dos eritrócitos e da hemossiderina que os acompanham é muito variável. Diferentes graus de diferenciação podem frequentemente ser encontrados no mesmo angiossarcoma. A epiderme pode ser normal, atrófica, ou ulcerada. A extensão da diferenciação dos angiossarcomas cutâneos não é tradicionalmente considerada como tendo um impacto no prognóstico, e assim, ao contrário de outros sarcomas, o grau histológico não é tido em conta para a encenação.39 Esta abordagem pode mudar no futuro, uma vez que um estudo recente com a maior série de angiossarcomas cutâneos e de tecidos moles publicado até à data, com 821 pacientes, desenvolveu um modelo de prognóstico para o angiossarcoma superficial que incluía o grau histopatológico.25
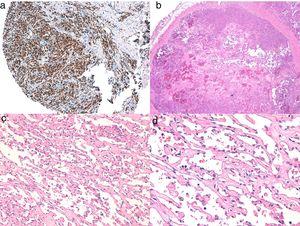 br>>>/div>
br>>>/div>
A, Imunohistoquímica do angiossarcoma cutâneo que é positivo para ERG (tipicamente com um padrão nuclear). B, Angiossarcoma com predominância de áreas com padrão vasoformativo. C, D, Imagens detalhadas de células endoteliais neoplásicas, que neste caso são proeminentes mas sem atipias notáveis.
Estudo de angiossarcoma deve ser completado com um painel imuno-histoquímico que inclui um painel básico para tumores de células fusiformes (CD31, pancytokeratins, S110, e actin) e marcadores vasculares adicionais (CD34, erythroblast transformation-specific related gene , podoplanin). Alguns casos de angiossarcoma com predominância de células epitelioides podem ser positivos para citoqueratinas; no entanto, a positividade para marcadores vasculares como CD31, ERG, e/ou podoplanina pode excluir o carcinoma indiferenciado. Nos últimos anos, tem sido demonstrado que muitos angiossarcomas têm amplificação/sobreexpressão MYC. Na maioria dos estudos, a amplificação de MYC encontra-se entre 50% e 100% dos angiossarcomas secundários, mas não geralmente nos idiopáticos,40-42 mas a amplificação ou sobreexpressão de MYC também foi detectada em alguns angiossarcomas idiopáticos.43,44 Contudo, quase todos os estudos demonstraram a ausência de amplificação ou sobreexpressão da MYC em proliferações vasculares atípicas pós-comercialização, e assim a positividade num caso duvidoso de proliferação vascular na área irradiada quase exclui o angiossarcoma.
p>A origem do angiossarcoma nos vasos sanguíneos ou linfáticos é sujeita a debate. A expressão de CD31 ou CD34 é maior nas áreas mais diferenciadas, mas em nenhum dos casos é constante.45 Os marcadores imunohistoquímicos relativamente específicos para vasos linfáticos, tais como podoplanina, D2-40, LYVE-1, e PROX-1, são geralmente positivos em angiossarcoma cutâneo, frequentemente expressos com um padrão imunohistoquímico misto de vasos sanguíneos endoteliais e vasos linfáticos endoteliais.38,46,47 Embora excepcionais, foram relatados casos de angiossarcoma cutâneo que expressam a proteína S-100,48 ou marcadores neuroendócrinos.49Estágio
Embora não existam directrizes para a gestão do angiossarcoma cutâneo, dado que o local mais frequente de metástase é o pulmão50 seguido pelos gânglios linfáticos,29,51 a tomografia computorizada toracoabdominal é geralmente recomendada após o diagnóstico patológico. Este estudo de imagem deve incluir a região cervical no caso de um angiossarcoma da cabeça e pescoço e a pélvis no caso de angiossarcomas abdominopelvícos pós-tradiação. A presença de disseminação regional ou distante não é incomum com os angiossarcomas, e é relatada em 30% e 10% dos casos, respectivamente.29
Não há estadiamento de TNM específico para os angiossarcomas, pelo que é utilizado o estadiamento de TNM para os sarcomas de tecido mole da classificação do Comité Misto Americano do Cancro, adaptado aos angiossarcomas. No caso dos angiossarcomas, como o grau histológico não é actualmente considerado como tendo impacto no prognóstico, os graus IA eIIA são agrupados em alguns estudos com os graus IB eIIB, que noutros sarcomas são diferenciados apenas pelo grau histológico.
Tratamento
O único tratamento potencialmente curativo no angiossarcoma cutâneo é a cirurgia, embora a cura ocorra apenas esporadicamente. Contudo, em casos inoperantes ou metastáticos, a radioterapia e/ou quimioterapia têm um papel paliativo reconhecido. Além disso, alguns estudos recentes incluíram também a radioterapia como adjuvante da cirurgia em casos localizados de angiossarcoma,50,51 e alguns autores recomendaram mesmo a irradiação de gânglios linfáticos regionais,52 mas esta não é uma prática comum. O problema da cirurgia no angiossarcoma cutâneo é por vezes o carácter multicêntrico e a fraca delimitação clínica noutros, bem como o facto de serem frequentemente diagnosticados quando as lesões excedem 5 cm de tamanho. Para além destes factores, os doentes são frequentemente idosos, o que torna mais difícil a obtenção de margens cirúrgicas adequadas. Em geral, se as características tumorais e o estado geral do doente o permitirem, o tratamento do angiossarcoma cutâneo é a excisão cirúrgica com margens suficientes. O mais aceite é a cirurgia com uma margem de 3 cm em relação aos limites clinicamente apreciáveis.53 A profundidade da margem não está bem estabelecida, dado que se trata de um sarcoma dérmico, embora pareça razoável atingir a fáscia sem os excisar. Nos casos mais infiltrativos, no entanto, o músculo deve ser incluído para alcançar margens claras. Margens com envolvimento de angiossarcoma demonstraram ser um factor de mau prognóstico em vários estudos.28,30,51 No caso do envolvimento de mama, a maioria dos estudos sugere mastectomia total ou excisões mais ou menos extensas de pele irradiada. Em casos complexos, o mapeamento prévio de biopsias pode ajudar no planeamento pré-cirúrgico. Sempre que possível, em oncologia cutânea, é preferível o encerramento directo, enxertos, ou encerramentos de segunda intenção para facilitar o seguimento e não mascarar possíveis recidivas locais com reconstrução cirúrgica, mas isto pode ser difícil ou impossível após excisões mais radicais de angiossarcomas mamários que requerem mastectomia total com irradiação de toda a pele. No caso de angiossarcomas causados por linfedema, um estudo analisou relatórios de 160 pacientes com síndrome de Stewart-Treves e encontrou ausência de benefício de amputação em comparação com a cirurgia radical (com 2 ou 3 cm de margem) nestes casos, pelo que a amputação de membros não parece justificar-se em tais angiossarcomas.54
Em casos em que a cirurgia é impossível, ou seja, lesões multicêntricas ou extensas, ou aquelas que afectam áreas que complicam a cirurgia, a radioterapia é o tratamento de escolha.55 A dose de radioterapia para o angiossarcoma cutâneo é normalmente 60 Gy distribuídos em 20 sessões de 3 Gy cada. Quando usado como adjuvante da cirurgia, as doses são semelhantes, excepto quando indicado para angiossarcoma pós-cirúrgico, em que a dose será normalmente mais baixa (45-50 Gy).
O único papel da quimioterapia no angiossarcoma é como tratamento paliativo, reservado para lesões recidivantes ou metastáticas não passíveis de cirurgia. Um papel neoadjuvante também foi recentemente proposto para quimioterapia antes da cirurgia em locais periorbitais.56 As quimioterapias mais utilizadas no angiossarcoma são o docetaxel,57,58 paclitaxel,59 e a doxorrubicina lipossomal,60 mas as actuais directrizes NCCN também incluem vinorelbina, sorafenibe, sunitinibe, e bevacizumab, embora os resultados com estes últimos 3 agentes antiangiogénicos tenham sido decepcionantes. A combinação com bloqueadores beta nesta fase do tratamento paliativo pode ser de algum benefício para o paciente.61,62
Follow-up
Não existem directrizes padrão para o seguimento de angiossarcomas cutâneos. No nosso grupo, temos um acompanhamento clínico rigoroso, com check-ups de 3 em 3-6 meses durante os primeiros anos, e depois check-ups anuais durante 10 anos. Nestas visitas, examinamos toda a pele e apalpamos os gânglios linfáticos territoriais correspondentes. Pelo menos uma vez por ano, realizamos uma análise laboratorial e um estudo de tomografia computorizada thoracoabdominopelvic.
Conflitos de interesse
Os autores declaram que não têm conflitos de interesse.